Autores:jjjjjjj Paulo Cesar Naoum Flávio Augusto Naoum |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
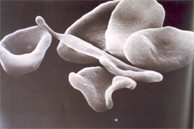
Introdução As talassemias beta são mais heterogêneas do que as do tipo alfa. Caracterizam-se por uma alteração quantitativa da síntese de globinas beta e são classificadas como talassemias beta zero (ou talassemia b 0) quando não há síntese de globinas, e talassemias beta mais (ou talassemia b+) quando há alguma taxa de síntese. Consequentemente as globinas alfa, que são sintetizadas normalmente, acumulam-se nos eritrócitos, durante a eritropoiese, causando agregação e precipitação. Os precipitados, formados em quantidades variáveis, danificam a membrana e destroem prematuramente essas células provocando a anemia (figura 7.11). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
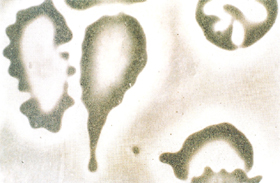
Figura 7.11: Lesões nas membranas de eritrócitos de pessoa com talassemia beta causadas pela precipitação de globinas alfa livres. Essas globinas livres causam a lipoperoxidação da dupla camada lipo-protéica dos eritrócitos talassêmicos, reduzindo o tempo de vida dessas células. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As supressões parcial e total das globinas beta, bem como das globinas delta e gama situadas no mesmo cromossomo 11, e que dão origem aos diferentes genótipos de talassemias b, d e g, estão representados na figura 7.12. De forma geral, os tipos mais comuns entre as talassemias beta são as heterozigoses: bA/b0, bA/b+ mediterrâneo, bA/b++ africano, bA/b+ silenciosa, bA/b0 com Hb A2 normal, e bA/b+ com Hb A2 normal. As talassemias beta homozigotas correspondentes são: b0/b0, b+/b+ mediterrâneo, b++/b++ africano, b+/b+ silenciosa, b0/b0 com Hb A2 normal, e b+/b+ com Hb A2 normal. A tabela 7.3 apresenta as principais características laboratoriais desses tipos de talassemias beta,nos estados de heterozigose e homozigose. A figura 7.13 representa esquematicamente as conseqüências laboratoriais da redução parcial ou total da síntese de globina beta, nas talassemias beta heterozigotas (ou menor), e nas talassemias beta homozigotas com redução parcial de síntese de globina beta (b+/b+ ou b0/b+) e redução total de globina beta (b0/b0), ambos são clinicamente caracterizados como talassemia beta maior. O modo de herança das talassemias, assim como de outras alterações genéticas da hemoglobina, é autossômico, e o termo dominante ou recessivo é difícil de ser aplicado, porque alguns heterozigotos apresentam claros distúrbios clínicos, ao passo que outros não. No entanto, a talassemia beta é considerada de herança autossômica recessiva, porque são necessários dois genes anormais da globina beta para produzir o fenótipo clinicamente detectável. Recentemente, no entanto, formas dominantes de talassemia beta têm sido identificadas, as quais resultam em fenótipos de talassemia intermédia para portadores de um único gene alterado. Com a utilização de técnicas de biologia molecular foi possível a identificação de aproximadamente 180 tipos diferentes de talassemias beta, cujas diversidades estão relacionadas com os graus de lesões no gene beta, podendo inclusive atingir os genes delta, pseudogene beta-1, os genes gama alanina e gama glicina e até o gene embrionário épsilon. A figura 7.14 representa por meio de traços pretos a intensidade de lesão por deleção sofrida pelos genes do complexo (b, d, gA, gG, wb1 e e), em alguns casos bem conhecidos de talassemia beta. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
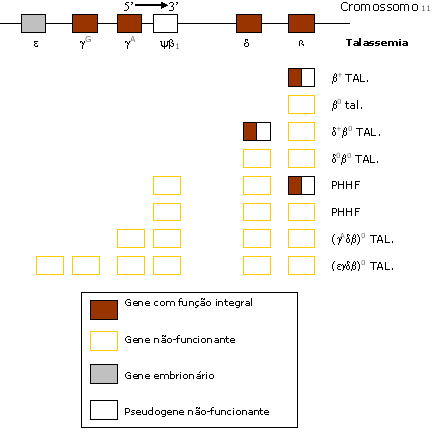
Figura 7.12: Representação esquemática de alguns genótipos de talassemia beta. Os genótipos b+ tal. e b0 tal. são as formas mais freqüentes seguidas de PHHF (persistência hereditária de Hb Fetal). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
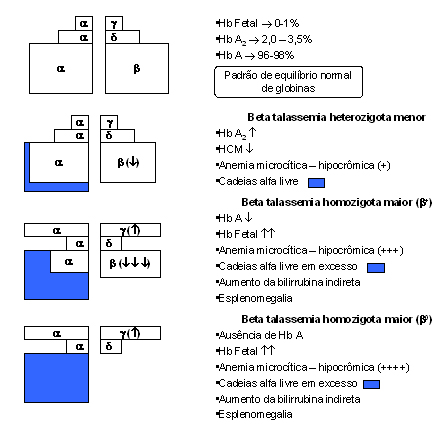 Figura 7.13: Relação entre quantidade
de globinas alfa livre, níveis de Hb A, Hb A2 e
Hb Fetal, e gravidade do quadro clínico em pacientes com talassemia
beta menor e maior.
Figura 7.13: Relação entre quantidade
de globinas alfa livre, níveis de Hb A, Hb A2 e
Hb Fetal, e gravidade do quadro clínico em pacientes com talassemia
beta menor e maior. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 7.3 – Principais características laboratoriais dos tipos mais comuns de talassemia beta. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
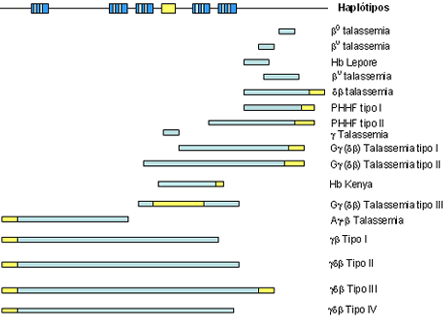
Figura 7.14: Esquema representativo do complexo gênico beta, relacionando os diferentes tipos de deleção que atingem os genes b, d, gA, gG, wb1 e e, as formas de talassemia beta. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fisiopatologia
O processo fisiopatológico da talassemia beta está muito relacionado com o desequilíbrio que se verifica entre as sínteses de globina alfa e beta. Com a síntese de globina beta afetada, por diminuição parcial (b+) ou bloqueio total (b0), a relação a/b supera o valor de equilíbrio que é de 1,0. A globina alfa, que não teve sua síntese alterada, apresenta produção normal, e como não há globina beta suficiente para formar tetrâmeros a2b2 ocorrerá a presença de globinas alfa livres, cuja intensidade é proporcional à piora do quadro clínico do portador, seja recém-nascido, ou com idade acima de 6 meses, conforme ilustra a figura 7.15. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
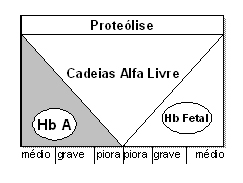
Figura 7.15: Representação gráfica do quadro clínico do paciente com talassemia maior (médio, grave e piora do quadro) relacionados com a quantidade de Hb A e Hb Fetal. Quanto maior a concentração de Hb A ou de Hb Fetal em g/dL o quadro melhora. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fisiologicamente o excesso de globinas (ou cadeias) alfa livres se instabiliza e se precipita sob forma de corpos de inclusão nos eritroblastos. Essa precipitação provoca situações patológicas celulares, conforme o local de sua ocorrência. Quando se dá na medula óssea, observa-se uma seqüência de fenômenos que se iniciam pela peroxidação dos lipídeos da membrana eritrocitária e geração de espécies ativadas de oxigênio, os radicais oxidantes ou radicais livres. A célula com baixa hemoglobinização é particularmente sensível a esse tipo de agressão tóxico-oxidante, pois a membrana lesada permite a perda de potássio e adenosina-tri-fosfato (ATP), tornando o eritrócito rígido, sem o poder natural da deformabilidade, e como conseqüência dificulta sua saída da medula óssea para o sangue periférico. Por outro lado, as células que têm maior hemoglobinização, mesmo que seja pela presença de Hb Fetal, apresentam-se com menores graus de lesões e, portanto, maior período de vida. Ainda em nível de células eritroblásticas, a precipitação de globinas alfa causa o bloqueio da síntese de DNA com conseqüente interrupção da síntese de globinas. O somatório das situações anteriormente mencionadas: precipitação de globina alfa, rigidez celular e lesão do DNA, provoca a eritropoiese ineficaz que é responsável pela situação de anemia e pelo aumento da absorção do ferro. Quando os eritrócitos com os corpos de inclusões compostos por globinas alfa atingem o sangue periférico, sua rigidez, associada às lesões na membrana eritrocitária, contribui para o seqüestro dessas células durante a circulação nos sinusóides esplênicos. O resultado dessa atuação fisiopatológica que se verifica nos doentes talassêmicos é a anemia hemolítica, com aumento da concentração da bilirrubina indireta e da esplenomegalia. A esplenomegalia pode resultar em hipereslenismo, pelo aumento da função do baço, levando a um processo mais abrangente de destruição das células do sangue, gerado a leucopenia e a plaquetopenia. A leucopenia pode contribuir par a instalação de infecções, constituindo-se em importante causa dos óbitos verificados em doentes com talassemia beta maior. A plaquetopenia induz, por sua vez, o derramamento de sangue nasal (epistaxes). As conseqüências provenientes da eritropoiese ineficaz, hemólise e hiperesplenismo resultam em anemia grave com anoxia, cardiopatias que são importantes causas de óbito na talassemia maior, hipermetabolismo com emagrecimento, febre, aumento do nível de ácido úrico e gota, e a necessidade de transfusões repetidas de sangue. Essas transfusões, que se tornam periódicas para os talassêmicos doentes, causam o acúmulo de ferro e da ferritina, podendo ocorrer hepatopatias graves por hepatite transfusional, além de doenças transmissíveis em transfusões, notadamente a AIDS e hepatite C, e esporadicamente doença de Chagas e Sífilis. Devido à deficiência significativa de globina beta, a formação de hemoglobina fica por conta das globinas alfa e gama, resultando a elevação percentual ou relativa da Hb Fetal. Embora, com baixa concentração corpuscular, a Hb Fetal com sua alta afinidade pelo oxigênio contribui para a anoxia tecidual. A anoxia tem efeitos maléficos ao organismo, predispondo-o às infecções, ao aparecimento de úlceras nas pernas e à interferência na secreção de eritropoietina que promove a hiperplasia eritróide. A hiperplasia eritróide causa o aumento da absorção do ferro, com seu acúmulo sob forma de ferritina. Nesse caso específico de acúmulo de ferro o doente talassêmico grave pode ter várias conseqüências patológicas desde que não utilize de processos de eliminação do ferro. Outras situações fisiopatológicas importantes se devem à diminuição do nível de folatos, que promove um quadro hematológico em sangue periférico similar ao da anemia megaloblástica, e as lesões ósseas determinam a presença de deformidades do crânio, maxilar e face, baixa estatura e a presença de massas extra-ósseas, ou metaplasias mielóides, com formações tumorais no mediastino e retroperitônio, ou acentuada esplenomegalia e hepatomegalia. Finalmente, o aumento da bilirrubina indireta, especialmente na talassemia beta maior, produz a excreção de dipirróis pela urina e litíase biliar. As figuras 7.16, 7.17 e 7.18 mostram de forma esquemática a seqüência dos eventos fisiopatológicos. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
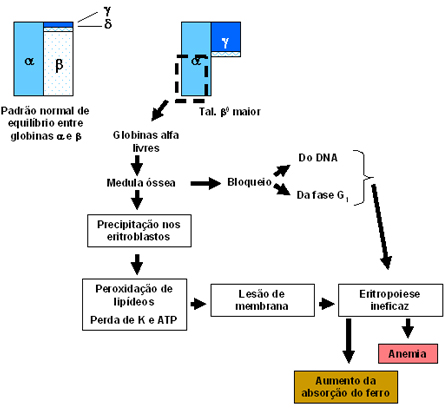
Figura 7.16: Conseqüências fisiopatológicas causadas pelo despareamento de globinas alfa livres. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
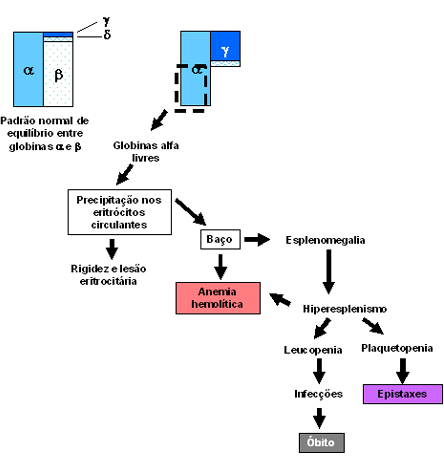 Figura 7.17: Conseqüências fisiopatológicas
causadas pelo despareamento de globinas alfa livres.
Figura 7.17: Conseqüências fisiopatológicas
causadas pelo despareamento de globinas alfa livres. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
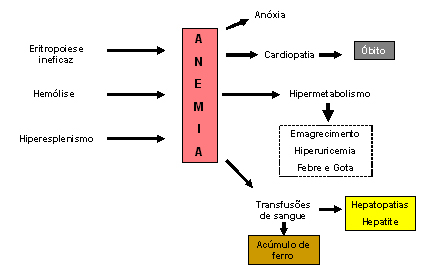 Figura 7.18: Intercorrências comuns na talassemia
beta maior.
Figura 7.18: Intercorrências comuns na talassemia
beta maior. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Talassemia beta homozigota (maior)
O primeiro relato científico
da talassemia beta homozigota foi realizado por um pediatra americano,
o Dr. Thomas B. Cooley, que juntamente com sua colega Pearl Lee
descreveu, em 1925, os achados hematológicos e clínicos
efetuados em quatro crianças que apresentavam anemia grave
com aumento do baço e deformidades dos ossos da face e
do crânio. Destacaram um fato importante: as crianças
tinham origem ancestral da região do mar Mediterrâneo,
por serem de descendências italiana e grega. A partir desse
relato, essa síndrome foi denominada por anemia de Cooley
& Lee, mas constantemente referida apenas como anemia de Cooley.
Alguns anos depois, devido à alta prevalência de
relatos similares, principalmente na Itália e na Grécia,
e também no Líbano, Tunísia, Argélia
etc., esses casos de anemias graves passaram a ser conhecidos
como Anemia do Mediterrâneo. Mais tarde, durante o Congresso
Internacional de Hematologia de 1940, um grupo de cientistas optou
pelo termo talassemia major, onde, em grego,
thalassa significa mar, e aima,
doença do sangue. A adjetivação da gravidade
inicialmente foi caracterizada pela palavra major,
que significa maior. Posteriormente, com o aprofundamento dos
estudos genéticos, passou-se a conhecer melhor as formas
de transmissões hereditárias e os defeitos dos genes,
e a forma grave foi denominada de talassemia beta homozigota;
a talassemia minor ou menor, de heterozigota;
e a talassemia intermédia foi definida por uma classificação
muito mais clínica do que genética ou laboratorial. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
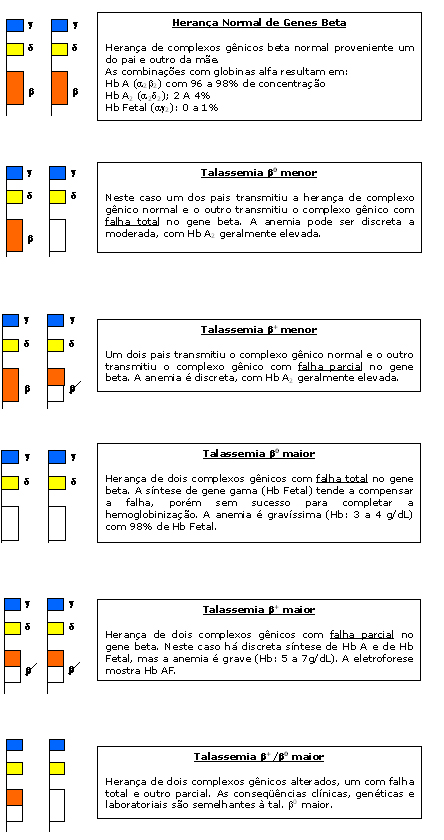 Figura 7.19: Representação gráfica
de diversas possibilidades de falhas no gene beta do complexo gênico
b, d e g
como resultado de talassemias beta menor e maior.
Figura 7.19: Representação gráfica
de diversas possibilidades de falhas no gene beta do complexo gênico
b, d e g
como resultado de talassemias beta menor e maior. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Muitos dos doentes afetados morrem na infância ou na adolescência, podendo, entretanto, alcançar a terceira década, conforme a atenção médica e terapêutica recebidas. Destaca-se, porém, que as principais causas de óbito são infecções ou insuficiências cardíacas, devido à deposição de ferro no miocárdio. O acúmulo de ferro é decorrente da extensa e prematura destruição dos eritrócitos, tanto daqueles que são continuamente produzidos para suprir a anemia hemolítica, quanto dos recebidos em transfusões sanguíneas freqüentes e necessárias, bem como da absorção gastrointestinal aumentada do ferro recebido pela dieta alimentar. O padrão de hemoglobinas nos pacientes com talassemia beta homozigota é variável, caracterizando-se pelo aumento de Hb Fetal, com concentrações que variam de 20 a 90%. A Hb A2 pode estar normal ou elevada e a Hb A aparece somente nos casos de deficiência parcial da síntese de cadeias beta. As crianças que não recebem tratamento adequado desenvolvem o quadro clínico típico da talassemia beta maior, que inclui, além da anemia grave, deformidades ósseas devidas à hiperplasia medular, hepatoesplenomegalia, pigmentação marrom da pele, distúrbios cardíacos e endócrinos, atraso no crescimento e na maturação sexual, infecções recorrentes e deficiência de ácido fólico. Por isso, para garantir a sobrevida dos pacientes, é necessário o tratamento contínuo que consiste em transfusões sanguíneas regulares, que mantêm um nível de hemoglobina adequado e diminue a atividade da medula óssea, e no uso de quelantes do ferro, que auxiliam a eliminação do excesso desse metal no organismo. As tabelas 7.4 e 7.5 resumem as principais características laboratoriais que são comuns em pacientes com talassemia beta homozigota ou maior. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 7.4: Sinopse das alterações laboratoriais em doentes com talassemia beta maior. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Talassemia
beta homozigota (menor)
O estado heterozigoto da talassemia beta é caracterizado geneticamente pela herança de um único componente alterado (figura 7.19). Nas formas b0 e b+, a redução da taxa de síntese da globina beta é menor, mas o suficiente para causar discreto grau de anemia microcítica e hipocrômica com aumento de resistência osmótica dos glóbulos vermelhos. São indistinguíveis por exames laboratoriais de rotina (tabela 7.3), entretanto, com a utilização de técnicas de síntese de cadeias ou de biologia molecular com sondas específicas de DNA, podem-se diferenciar esses heterozigotos. Muitas vezes, a doença é mal diagnosticada e os pacientes são tratados inadequadamente, como se apresentassem anemia por deficiência de ferro. Laboratorialmente, as formas de talassemia b0 ou b+ caracterizam-se pelo aumento de Hb A (fig.7.20) cuja concentração varia de 4 a 7%, alterações morfológicas dos eritrócitos identificados especialmente por microcitose e hipocromia com muitos esquisócitos, dacriócitos e pontilhados basófilos (fig. 7.21), resistência osmótica aumentada na solução de NaCl a 0,36%, diminuição da hemoglobina corpuscular média (HCM) e do volume corpuscular médio (VCM). A Hb Fetal pode estar normal ou discretamente aumentada. As manifestações clínicas, quando presentes, variam entre os diferentes grupos raciais, e entre elas podemos citar astenia, cansaço e baço palpável. A artrite também pode ser constatada na talassemia beta heterozigota. Os níveis de ácido fólico e vitamina B12 plasmáticas apresentam-se dentro dos limites normais em talassêmicos beta heterozigotos. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
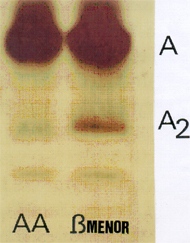
Figura 7.20: Hb A2 aumentada em portadores de talassemia beta menor, visualizada por meio de eletroforese alcalina (pH 8,5) em acetato de celulose em comparação com Hb AA e Hb A2 normal. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
De uma forma geral, a maioria dos portadores de talassemia beta, heterozigota apresentam padrões hematológicos que são coincidentes em 90% dos casos descritos na literatura: VCM: (61 a 73) fl HCM: (20 a 24) pg Hb A2: (4 a 7)% Formas atípicas de talassemia beta heterozigota podem ocorrer, das quais os principais tipos são os seguintes: Tipo 1 – talassemia beta heterozigota, com Hb A2 aumentada, VCM e HCM normais. Tipo 2 – talassemia beta heterozigota, com Hb A2 diminuída, Hb Fetal discretamente elevada, VCM e HCM diminuídas. Esses casos são suspeitos de talassemia heterozigota beta-delta. Tipo 3 – talassemia beta heterozigota, com Hb A2 normal e VCM e HCM diminuídos. Esses casos são suspeitos da associação entre talassemia alfa e beta heterozigotas. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
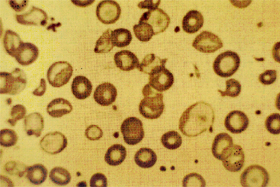
Figura 7.21: Morfologia eritrocitária típica de talassemia beta menor. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Talassemia
intermediária As formas clínicas denominadas por talassemia intermédia são aquelas resultantes de diferentes interações genéticas, cujos portadores apresentam quadro clínico mais ameno do que o da talassemia beta maior e não são dependentes de transfusão sanguínea. A talassemia beta intermédia pode decorrer da interação das talassemias alfa e beta, com redução concomitante e significativa de ambas as cadeias globínicas, o que diminui o número de cadeias desemparelhadas e propicia uma redução na taxa de destruição dos eritrócitos em comparação com as formas graves de talassemias. Entretanto a forma mais prevalente de talassemia beta intermédia se deve a lesões do tipo b+ ( b+/ b+), cujo diagnóstico laboratorial somente é feito por biologia molecular. A talassemia beta intermédia pode decorrer também de manifestações da talassemia beta com alguns tipos de hemoglobinas variantes, particularmente a Hb E, Hb S e Hb C. Interações entre talassemia
beta e Hb variantes
As formas interativas mais freqüentes entre talassemias beta e hemoglobinas variantes, no Brasil, são Hb S/beta talassemia e Hb C/beta talassemia. Há três formas principais de Hb S/beta talassemia: (a) Hb S/b0 talassemia, (b) Hb S/b+ talassemia tipo Mediterrâneo, (c) Hb S/b++ talassemia tipo Negro. A forma Hb S/b0 talassemia apresenta-se clinicamente parecida com a anemia falciforme, ou Hb SS, mas seus eritrócitos mostram os valores de HCM e VCM diminuídos. Na análise eletroforética a concentração de Hb S é variável entre 80 e 90%, a Hb Fetal está aumentada (até 10%), há ausência de Hb A, e a Hb A2 está constantemente elevada. Para se efetuar um diagnóstico laboratorial seguro é importante a realização de exames nos pais dos portadores, que certamente se apresentarão como sendo um deles portador de talassemia b heterozigota e outro de Hb S. Para melhor entendimento veja a figura 6.55, no capítulo 6 “Doença falciforme” – Diagnóstico laboratorial das doenças das células falciformes, deste site. A forma de Hb S/ talassemia b+ Mediterrâneo é similar em gravidade à anemia falciforme, excetuando-se com relação às crises de falcização, que ocorrem com menor freqüência, apresentando, entretanto, um hiperesplenismo que causa anemia mais proeminente. As alterações nos eritrócitos são características das talassemias e na análise eletroforética observa-se a presença de Hb A (15 a 30%), bem como Hb S, sempre com maior concentração que a Hb A. A Hb Fetal e Hb A2 podem estar elevadas, sem ser regra geral. Para melhor entendimento veja as figuras 6.56 e 6.57, no capítulo 6 “Doença falciforme” – Diagnóstico laboratorial das doenças das células falciformes, deste site. A forma Hb S/talassemia b++ tipo Africano é moderada, os portadores são menos anêmicos do que aqueles com a forma Mediterrânea. Os índices hematimétricos são parecidos com aqueles observados em talassêmicos heterozigotos. A análise eletroforética mostra a concentração de Hb A variável entre 25 e 30%, Hb S com níveis quantitativos superiores ao da Hb A, e hemoglobinas A2 e Fetal com valores elevados. As interações envolvendo Hb C e talassemia beta não diferem muito em relação à explanação anterior a respeito da Hb S. Talassemia beta-delta
Essa talassemia é resultante da deficiência de ambas globinas beta e delta, que está associada à hemoglobinização deficiente dos eritrócitos. A talassemia beta-delta no estado de homozigose produz quadro clínico semelhante ao verificado na talassemia intermédia. A Hb Fetal constitui 100% da concentração total da hemoglobina, devido à ausência de síntese das hemoglobinas A e A2. A análise citológica dos eritroblastos, corados com solução supravital, mostra a presença de agregados de cadeias alfa, enquanto a fragilidade osmótica e a HCM estão diminuídas. A forma heterozigota, por sua vez, é caracterizada por níveis normais de Hb A2, a Hb Fetal apresenta-se com valores variáveis entre 5 a 20%, a anemia é discreta e a HCM está diminuída na maioria dos portadores. A Hb Fetal está heterogeneamente distribuída entre os glóbulos vermelhos quando analisada em esfregaço submetido ao teste de eluição ácida, conforme mostra a figura 7.22. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
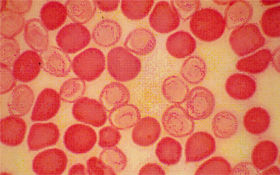
Figura 7.22: Distribuição heterogênea de Hb Fetal intra-eritrocitária. Esse teste citológico é usado para diferenciar o aumento da concentração de Hb Fetal que ocorre na persistência hereditária de Hb Fetal (distribuição homogênea) das talassemias beta maior e intermédia (distribuição heterogênea). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Associação
entre talassemias beta e beta-delta A heterozigose dupla para talassemias beta e beta-delta com Hb Fetal elevada é mais freqüente na Grécia. Os portadores dessa forma de talassemia têm anemia de gravidade intermediária, geralmente compensada sem transfusões. Entretanto, os sinais clínicos e a manifestação da anemia variam consideravelmente, e alguns pacientes podem ter hepatoesplenomegalia, anemia e osteoporose. Os pacientes padecem, ainda de predisposição à anemia hemolítica auto-imune. Os resultados hematológicos são similares àqueles encontrados na talassemia beta homozigota, com graves alterações eritrocitárias, diminuição da HCM e da fragilidades osmótica, corpos de inclusão são compostos por agregados de cadeias alfa livre, alta concentração de Hb Fetal (50 a 90%) e Hb A2 normal ou diminuída (figura 7.23). A distribuição da Hb Fetal intracelular é geralmente homogênea. O diagnóstico é confirmado por exames nos pais, um deles é heterozigoto para talassemia beta e outro para variante beta-delta com Hb Fetal elevada. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
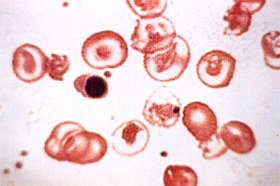
Figura 7.23: Esfregaço de sangue de paciente com anemia grave, típica de beta maior, mas que análises laboratoriais revelaram ter genótipo b0d/b0d. Hipocromia acentuada, eritroblastos, corpos de Howell-Jolly, etc. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Talassemia
delta Desde que a Hb A2 representa somente uma menor fração da hemoglobina total, o desequilíbrio da síntese de cadeia delta não tem efeito significativo na morfologia dos glóbulos vermelhos e não tem importância clínica, a menos que esteja associada com outra hemoglobina anormal. Na talassemia delta heterozigota espera-se uma diminuição da Hb A2, enquanto nos homozigotos esta fração está totalmente ausente.
As talassemias beta constituem um grupo de
alterações genéticas da síntese de
hemoglobina extremamente diverso e que resulta na diminuição
da globina beta. Clinicamente há grande variabilidade com
relação a sintomas e manifestações,
e essas condições são resultantes de fatores
genéticos e adquiridos. A variabilidade clínica
e hematológica sugere heterogeneidade genética,
que é confirmada atualmente pela grande gama de mutações
e alterações gênicas que originam as talassemias
beta e cuja classificação pode ser realizada por
análise de DNA. O complexo gênico das cadeias do
tipo beta compreende uma extensão de aproximadamente 50Kb,
incluindo os cinco genes funcionais e o pseudogene, e estão
arranjados na seqüência 5’-3’, na mesma
ordem em que são ativadas durante as fases do desenvolvimento
humano (ver figura 3.5 do capítulo 3 ‘Hemoglobinopatias”
– Síntese Genética, deste site).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 7.5: Análises laboratoriais para o diagnóstico das talassemias heterozigotas. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Primeiro Nível Índices eritrocitários:
atualmente são determinados quase exclusivamente com contadores
automáticos de células do sangue. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
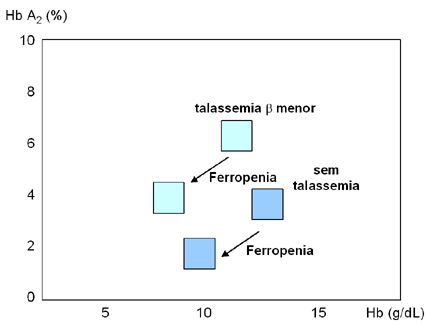 Figura 7.24: Gráfico relacionado a influência
da anemia ferropriva na dosagem de Hb A2, tendo como exemplo
um caso com talassemia beta menor e outro sem talassemia.
Figura 7.24: Gráfico relacionado a influência
da anemia ferropriva na dosagem de Hb A2, tendo como exemplo
um caso com talassemia beta menor e outro sem talassemia. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
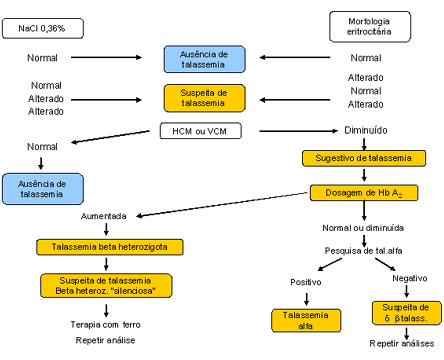 Figura 7.25: Sinopse em forma de esquema dos testes
seletivos e específicos para talassemia.
Figura 7.25: Sinopse em forma de esquema dos testes
seletivos e específicos para talassemia. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Segundo Nível Análise de hemoglobina com técnicas específicas: a aplicação da técnica de eletroforese por isoeletrofocalização ocorre em casos muito específicos, com o objetivo de separar hemoglobinas variantes com fenótipos talassêmicos. O melhor exemplo da aplicação dessa técnica se refere à Hb Knossos, uma hemoglobina estruturalmente anormal, que ao se associar à talassemia beta heterozigota provoca a redução na síntese de RNAm para a globina beta. Essa hemoglobina variante não se evidencia por meio de técnicas comuns de eletroforese, necessitando da isoeletrofocalização. Entre as técnicas específicas incluem pesquisa de corpos de Heinz e raramente síntese de globina. Terceiro Nível Análise do DNA: o estudo do DNA é realizado por tecnologia molecular e tem sido aplicado no diagnóstico de talassemias que apresentam dificuldade de identificação pelas técnicas descritas nos primeiro e segundo níveis. É usada com muita frequência, também para estabelecer os pontos de mutações que ocorrem no complexo gênico beta, permitindo, dessa forma, a diferenciação molecular de diversos genótipos de talassemia beta. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||