Talassemia Alfa
|
|||||||||||||||||||||||||||||||||||||||
Introdução Em 1955, foi descrita pela primeira vez nos Estados Unidos e na Grécia uma nova hemoglobina rápida que denominaram de Hb H, com características instáveis, formando corpúsculos de inclusões nos eritrócitos e visualizados quando submetida à incubação com corantes vitais. Esses achados estavam associados às alterações morfológicas dos eritrócitos, desde que afastadas as possibilidades de que a anemia microcítica e hipocrômica fosse devido à deficiência de ferro. A identificação de que a Hb H era composta por tetrâmeros de globinas beta, realizada por técnicas de hibridização e por análises bioquímicas, sugeriu que se tratava de uma doença causada por defeito nos genes alfa. A Hb H tem afinidade ao oxigênio 10 vezes maior do que a Hb A, ausência de efeito Bohr e liga-se ao cromo mais rapidamente do que a Hb A. Os portadores dessa alteração molecular, denominada doença de Hb H, apresentavam anemia significativa, hipocromia, microcitose e poiquilocitose, diminuição da fragilidade osmótica, reticulocitose e presença de Hb H nos eritrócitos e em eletroforese.Em 1965, foi introduzida a técnica de avaliação da síntese de globina alfa e beta, por meio da utilização de reticulócitos incubados com leucina marcada radioativamente. A medida da quantidade de síntese de globina era avaliada pela relação alfa/beta, cujo valor médio se situa em torno de 1,0. Ao analisarem os casos classificados como talassêmicos alfa, os pesquisadores observaram que a relação alfa/beta era menor do que 1,0 e concluíram, por isso, que na talassemia alfa ocorria um decréscimo na síntese de globina alfa. Em 1974, o Professor Lehmann da Universidade de Cambridge, considerado um dos mais conceituados pesquisadores em hemoglobinopatias, obteve com uma simples observação a resposta sobre o número de genes alfa presentes numa pessoa. Relacionando os casos de hemoglobinas variantes com alterações por trocas de aminoácidos na globina alfa, verificou que todas as Hb variantes de globina alfa apresentavam concentrações inferiores a 25%, quando associadas com a Hb A. Esse fato levou-o a concluir que cada pessoa possui quatro genes alfa, sendo dois genes em cada um dos cromossomos 16 onde cada um sintetiza cerca de 25% da globina alfa. Em 1978, com utilização de métodos de biologia molecular, ficou comprovada a hipótese do Professor Lehmann. Na década de 80, os estudos moleculares realizados com os genes de globina alfa revelaram que vários defeitos genéticos podem provocar a talassemia alfa, e que dependendo da extensão da lesão do gene, a síntese de globina alfa apresenta diferentes intensidades de decréscimos, incluindo a ausência total de síntese. Como resultado desse desequilíbrio entre as sínteses de globina alfa e beta, a globina beta continua sendo sintetizada normalmente e, por isso, a “sobra” de globinas beta livres se juntam para formarem tetrâmeros de globinas b 4, resultando a Hb H. Quanto maior a queda de síntese de globina alfa, maior será também a concentração de Hb H. Nos recém-nascidos, a diminuição da síntese de globina alfa afeta sua relação com a globina gama normalmente sintetizada. Assim, formam-se tetrâmeros de globina g 4, resultando na Hb Bart’s. É importante destacar que as talassemias alfa podem ter duas causas de origem: hereditária e adquirida. Evidentemente as formas hereditárias são as mais comuns e atingem, pelo menos, 20% da população brasileira dos quais 17% são assintomáticos e com valores hematimétricos (Hb, Ht, VCM e HCM) normais; 3% tem discretos graus de anemia microcítica e hipocrômica, e 1:5.000 pessoas é portadora da doença de Hb H). As formas adquiridas são secundárias a um processo patológico primário, por exemplo: doenças linfo e mieloproliferativas, anemia sideroblástica, entre outras. Classificação As talassemias alfa são diferenciadas
e classificadas de acordo com o número de genes alfa lesados,
com a importante observação de que o grau de lesão
pode ser variável, afetando o gene parcial ou totalmente.
De uma forma geral, representa-se uma pessoa sem talassemia alfa
com seus quatro genes alfa funcionantes (a
,a /a ,a
), sendo dois genes alfa de um cromossomo 16 e dois do outro cromossomo
16, provenientes um do pai e outro da mãe. |
|||||||||||||||||||||||||||||||||||||||
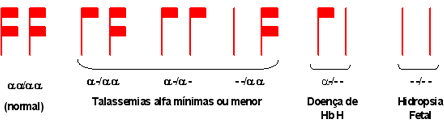
Esquema 1 - Tipos de lesões que causam talassemia alfa. |
|||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||
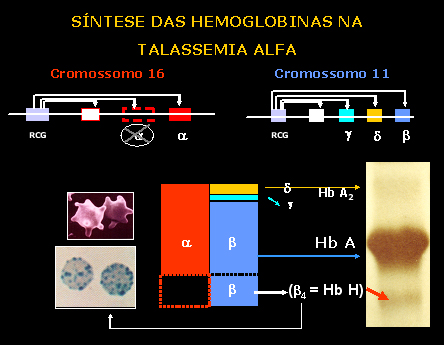
Esquema 2 - Representação da lesão do gene alfa no cromossomo 16 e suas conseqüências. |
|||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||
As talassemias alfa pela sua diversidade de manifestações clínicas e laboratoriais são também conhecidas por síndromes alfa talassêmicas. Essas variabilidades podem ocorrer dentro de cada grupo étnico, dependendo da especificidade de mutações e de como se expressam. De uma forma geral, as síndromes alfa talassêmicas são classificadas em: portador “silencioso” (ou talassemia alfa mínima), traço alfa talassêmico (ou talassemia alfa menor), doença de Hb H (ou talassemia alfa intermédia) e hidropsia fetal. Portador “silencioso” – é o tipo mais comum entre as talassemias alfa e se deve à deleção de apenas um gene alfa (-, a /a ,a ). O portador desse tipo de talassemia é assintomático, e embora o volume corpuscular médio (VCM) se apresente como discretamente microcítico (VCM < 80), a morfologia eritrocitária é geralmente normal com microcitose em algumas células. A análise eletroforética de hemoglobina hemolisada com saponina a 1% pode revelar traços de Hb H que representam concentrações inferiores a 1%. Da mesma forma, se a análise for efetuada em sangue de cordão umbilical, ou recém-nascidos, a concentração de Hb Bart’s (g 4) situa-se entre 1 e 2%. A pesquisa intra-eritrocitária de Hb H, após 30-60 minutos de incubação do sangue com azul de cresil brilhante a 37°C, pode revelar uma célula positiva para cada 1.000 ou 2.000 pesquisadas. Entretanto, nem sempre que aparece o traço de Hb H na eletroforese, a pesquisa intra-eritrocitária de Hb H resulta positiva. O diagnóstico laboratorial do portador silencioso de talassemia alfa requer uma série de informações: discreta microcitose, com valores de Hb (g/dL) próximo do limite inferior da normalidade, não-responsiva ao tratamento com ferro, história familiar, e identificação da Hb H em pelo menos um dos testes: eletroforese ou pesquisa citológica. A prevalência média do portador silencioso para talassemia alfa é próximo de 17% na população brasileira. Traço alfa talassêmico – se deve à deleção de dois genes alfa (-,-/a ,a ) ou (-,a /-,a ). Os portadores, apesar de serem normais sob o ponto de vista clínico, reclamam de fraqueza, cansaço, dores nas pernas e palidez. Apresentam microcitose com alterações da morfologia eritrocitária e discreto grau de anemia (Hb: 11 a 13g/dL, VCM e HCM diminuídos). A análise eletroforética da hemoglobina hemolisada com saponina a 1% mostra a presença de Hb H com concentrações próximas de 2% (fig.7.1). |
|||||||||||||||||||||||||||||||||||||||
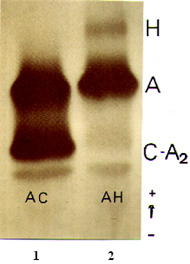
Figura 7.1 - Eletroforese de hemoglobinas em acetato de celulose pH 8,6. (1) portador de Hb AC; (2) portador de traço alfa talassêmico. |
|||||||||||||||||||||||||||||||||||||||
A pesquisa intra-eritrocitária de Hb H, após 30-60 minutos de incubação a 37°C com azul de cresil brilhante, permite a visualização de uma célula positiva para cada 250 a 500 pesquisadas (fig.7.2). |
|||||||||||||||||||||||||||||||||||||||
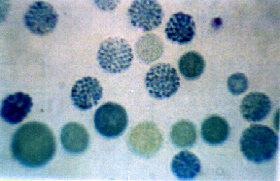
Figura 7.2 - Precipitados intra-eritrocitários de Hb H em sangue de portador do traço alfa talassêmico, incubado a 37°C com azul de cresil brilhante por 60 minutos. |
|||||||||||||||||||||||||||||||||||||||
Da
mesma forma que o caso anterior, a história clínica
do paciente e o estudo familiar são fundamentais para se
chegar ao diagnóstico do traço alfa talassêmico.
Sua prevalência na nossa população é
próxima dos 3%. A detecção do traço
alfa talassêmico é mais sensível de se realizar
em sangue de cordão umbilical ou em recém-nascidos
com um mês de idade, pois a Hb Bart’s apresenta-se com
concentrações variáveis entre 5 e 10%. Doença de Hb H – é causada pela deleção de três genes alfa (-,-/-,a ) . Essa patologia se expressa com uma forma moderadamente grave de talassemia, caracterizada por anemia microcítica e hipocrômica, hemoglobina total variável entre 8 e 11g/dL, aumento do baço e do fígado, e em alguns casos observa-se deformidades similares às que ocorrem na talassemia beta intermédia. A hemoglobina H separada por eletroforese alcalina, em sangue hemolisado com saponina a 1%, apresenta-se bem visível, pois sua concentração atinge até 20% (figura 7.3). A Hb H intra-eritrocitária é facilmente identificada pela sua presença em vários eritrócitos em um mesmo campo microscópico (figura 7.4). Em recém-nascidos, a Hb Bart’s apresenta-se com concentrações entre 20 e 30%. A doença de Hb H é rara no Brasil, apesar de vários relatos científicos provenientes de diferentes regiões do país. |
|||||||||||||||||||||||||||||||||||||||
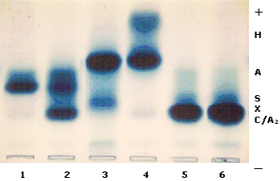
Figura 73. - Eletroforese de hemoglobinas em gel de agarose alcalina. O caso nº 4 é específico de doença de Hb H, com concentrações de 20% desta hemoglobina, 79% de Hb A e 1% de Hb A2. Os outros casos são: 1: Hb SS; 2: Hb SC; 3: Hb A + Hb Instável Koln; 5 e 6: Hb CC. |
|||||||||||||||||||||||||||||||||||||||
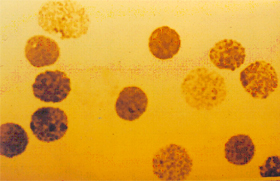
Figura 7.4 - Precipitados intraeritrocitários de Hb H na doença de Hb H. |
|||||||||||||||||||||||||||||||||||||||
Hidropsia Fetal – é a forma mais grave de todos os tipos de talassemias (alfa e beta), pois é uma forma letal. É uma situação comum no Extremo Asiático, sendo, entretanto, esporádica no Brasil. As crianças recém-nascidas afetadas pela deleção dos quatro genes alfa (-,-/-,-) apresentam anemia muito grave, com hemoglobina inferior a 7g/dL, eritroblastose fetal, edema, grande aumento do baço e do fígado, e morte com poucas horas após o nascimento. Eletroforeticamente, a concentração de Hb Bart’s, está entre 80 e 100%, e a Hb H entre 10 e 20%. A tabela 7.1 e a figura 7.5 apresentam um resumo dessas quatro condições básicas de talassemia alfa. |
|||||||||||||||||||||||||||||||||||||||
Tabela 6.17 – Diferenciação dos principais genótipos de Hb S relacionados aos valores de hemoglobina (Hb g/dl), volume corpuscular médio (VCM), reticulócitos e Hb Fetal. |
|||||||||||||||||||||||||||||||||||||||
P.I.E.: Pesquisa intra-eritrocitária.
*: Elevação do VCM devido a reticulocitose. |
|||||||||||||||||||||||||||||||||||||||
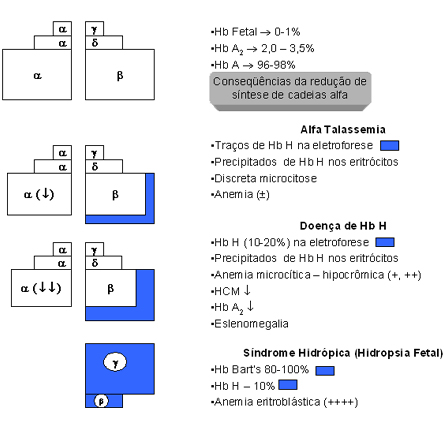 Figura 7.5 - Representação esquemática
da fisiopatologia da talassemia alfa, relacionando o traço
alfa talassêmico, a doença de Hb H e a hidropsia fetal
com a diminuição de síntese da globina alfa.
Figura 7.5 - Representação esquemática
da fisiopatologia da talassemia alfa, relacionando o traço
alfa talassêmico, a doença de Hb H e a hidropsia fetal
com a diminuição de síntese da globina alfa.
|
|||||||||||||||||||||||||||||||||||||||
Finalmente é importante destacar que as hemoglobinas H e Bart’s apresentam alta afinidade pelo oxigênio, tornando sua liberação para as células e tecidos muito lenta e dificultosa, e como resultado causa anoxia tecidual. Esse processo é mais grave e proporcional ao número de genes alfa afetados, conforme pode ser apreciado na figura 7.5 e tabela 7.1. Da mesma forma, os graus de hemólise e de anemia também estão na dependência da quantidade de Hb H precipitada nos eritrócitos. Base molecular das Talassemias Alfa O agrupamento de genes alfa está localizado no braço curto do cromossomo 16, e contém três genes ativos: o gene zeta da fase embrionária, e os genes a1 e a2 (vide esquema abaixo). A seqüência de bases nitrogenadas nos dois genes alfa é idêntica, podendo, entretanto, ocorrer algumas diferenças em situações anormais, por exemplo: o “crossing-over” desigual (fig. 7.6), com resultados desproporcionais em relação ao número de genes alfa por cromossomo 16. Atualmente se sabe que o gene a2 produz de duas a três vezes mais RNA mensageiro que o gene a1, mas a mensagem do gene a1 é traduzida com maior rapidez e, por isso, a diferença de síntese entre os genes a2 e a1 não é tão grande como se poderia esperar. A figura 7.7 mostra que cada gene alfa está localizado dentro de regiões de homologia, representadas pelas letras, X, Y e Z, e que são interrompidas por duas regiões curtas e não-homólogas, para cada espaço de 4,2Kb (entre os genes ya1 e a2) e 3,7Kb (entre os genes a2 e a1). É importante destacar que no agrupamento de genes alfa, representado pela seguinte proposição esquemática: |
|||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||
Há
duas regiões hipervariáveis, uma localizada entre
o gene z2 e o yz1,
o outro após o gene q1. Por essa
razão ocorrem consideráveis variações
na estrutura do agrupamento de genes alfa em pessoas consideradas
normais para a expressão de síntese de globina alfa.
Assim, embora a maioria das pessoas tenha quatro genes alfa (a
, a /a ,a
), cerca de 2% da população mundial tem cinco genes
(a , a ,a
/a ,a ). |
|||||||||||||||||||||||||||||||||||||||
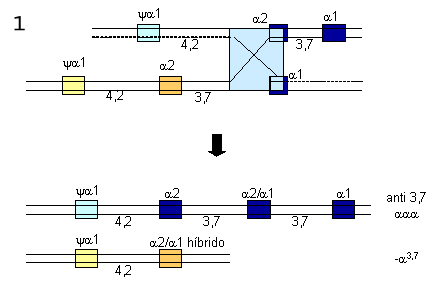
OU 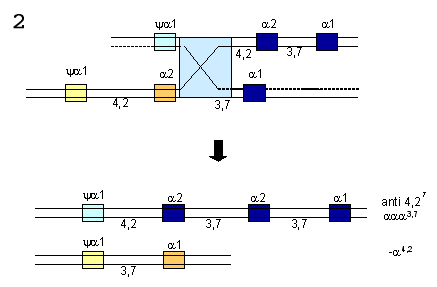 Figura 7.6 – “Crossing-over” desigual entre dois cromossomos 16. (1) inserção de três genes alfa (a2,a2/a1,a1) num dos cromossomos, e representado por aaaanti3,7. No outro cromossomo, restou apenas um gene híbrido (a1, a2), e representado por -a3,7. (2) da mesma forma esse processo genético pode ocorrer sem formação de genes alfa híbridos, e são representados por aaaanti 4,2 e -a4,2. |
|||||||||||||||||||||||||||||||||||||||
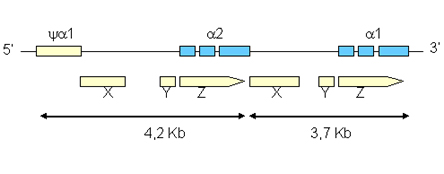 Figura 7.7 – Regiões homólogas
entre os espaços ya1
e a 2,
e entre os genes a 2
e a 1.
Essas regiões homólogas têm diferentes tamanhos
em números de bases nitrogenadas: 3,7Kb e 4,2Kb, e são
denominadas de X, Y e Z. Entre as regiões homólogas
X e Y, e entre Y e Z, localizadas no espaço de 4,2Kb, há
dois espaços que correspondem às regiões não-homólogas.
O raciocínio é o mesmo em relação às
regiões homólogas e não-homólogas localizadas
no espaço 3,7Kb. O tamanho das regiões homólogas
nos espaços 3,7 e 4,2 são iguais, enquanto os das regiões
não-homólogas são diferentes.
Figura 7.7 – Regiões homólogas
entre os espaços ya1
e a 2,
e entre os genes a 2
e a 1.
Essas regiões homólogas têm diferentes tamanhos
em números de bases nitrogenadas: 3,7Kb e 4,2Kb, e são
denominadas de X, Y e Z. Entre as regiões homólogas
X e Y, e entre Y e Z, localizadas no espaço de 4,2Kb, há
dois espaços que correspondem às regiões não-homólogas.
O raciocínio é o mesmo em relação às
regiões homólogas e não-homólogas localizadas
no espaço 3,7Kb. O tamanho das regiões homólogas
nos espaços 3,7 e 4,2 são iguais, enquanto os das regiões
não-homólogas são diferentes. |
|||||||||||||||||||||||||||||||||||||||
As mutações mais comuns que afetam as atividades de síntese de globina alfa podem ser por deleção e não-deleção. As deleções ocorrem particularmente por efeito de “crossing-over” desigual devido ao desalinhamento entre os cromossomos 16 durante o processo de meiose (fig. 7.6). O “crossing-over” entre as regiões homólogas do tipo Z, geralmente envolve mudanças entre os genes a1 e a2 deletando 3,7Kb de DNA, e deixando um simples gene a2, ou um gene alfa híbrido a1 / a2, num dos cromossomos. Esse defeito é conhecido por deleção –a37. No outro cromossomo com três genes alfa (aaa), o portador não é afetado, pois não se demonstrou até o presente nenhuma desvantagem seletiva, desde que associado a combinações, com os genes do tipo beta normais (g, d e bA). O “crossing-over” entre as regiões X não envolve genes, perdendo 4,2Kb, e por isso a mutação é conhecida por deleção –a4,2. O “crossing-over” envolvendo a região Y ainda não foi identificado. A nomenclatura das falhas de expressão do gene alfa está especificamente relacionada com a quantidade de produção de globina alfa. Denomina-se de talassemia a0, quando determinado gene alfa não sintetiza globina alfa, enquanto àqueles com síntese apenas reduzida refere-se como talassemia a+. Os haplótipos normais para globina alfa são representados por aa, indicando os genes a2 e a1, respectivamente. O indivíduo com produção normal de globina alfa tem seu genótipo representado por dois haplótipos aa (a, a /a, a ). Quando ocorre deleção do gene alfa, envolvendo um gene alfa (-a) ou os dois genes alfa (- -) é possível conhecer a extensão da lesão por estudos moleculares. A deleção -a3,7 indica o desaparecimento de 3,7Kb do complexo gênico. Quando o tamanho de uma deleção não está bem estabelecido, usa-se identificá-la pelo local de proveniência do portador, por exemplo: a representação - -Med descreve uma deleção de ambos genes alfa obtida de análise de material proveniente da região do mar Mediterrâneo. Talassemias a0 – até o presente 12 deleções foram descritas, envolvendo os dois genes alfa (a2 e a1), conforme mostra figura 7.8. Análises detalhadas sugerem quebras na região do agrupamento dos genes e pseudogenes do agrupamento alfa, similares estruturalmente aos que ocorrem nas translocações cromossômicas que originam algumas doenças malignas. Esse contraste com os defeitos -a3,7 e -a4,2 que são deleções comuns em diversas populações, os tipos a0 tem limitadas distribuições geográficas, e cada uma representa uma forma rara e incomum ocorrida por algum acidente genético. |
|||||||||||||||||||||||||||||||||||||||
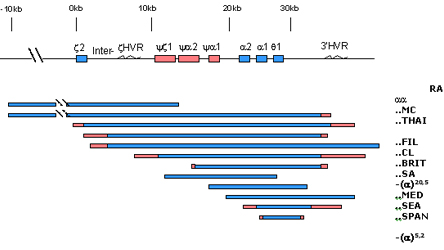 Figura 7.8 – Sumário esquemático dos principais
tipos de deleções no agrupamento de genes alfa, cujos
resultados se manifestam pela perda de expressão dos genes
alfa. Por exemplo: a deleção aaRA
atinge a região hipervariável (HVR) e o gene z
2, a deleção - -MC atinge
todos os genes do agrupamento.
Figura 7.8 – Sumário esquemático dos principais
tipos de deleções no agrupamento de genes alfa, cujos
resultados se manifestam pela perda de expressão dos genes
alfa. Por exemplo: a deleção aaRA
atinge a região hipervariável (HVR) e o gene z
2, a deleção - -MC atinge
todos os genes do agrupamento. |
|||||||||||||||||||||||||||||||||||||||
Talassemias a+ – é o defeito molecular mais comum, envolvendo a deleção de um dos genes alfa, conforme mostra a figura 7.1. O nível de expressão do gene alfa que não foi afetado exerce sua atividade de síntese que expressa diferentemente e proporcionalmente ao número de genes alfa ativos. Destaca-se, entretanto, que a avaliação mais precisa é efetuada pela medida de produção do RNAm específico para o gene alfa em ambos os cromossomo 16. São cinco deleções que ocorrem na talassemia a+: -a3,1, -a3,7111, -a3,7111, -a4,2 e -a3,13,5, todas com capacidade de reduzir a produção de globina alfa. As talassemias a+ por processo de não-deleção se deve, na maioria dos casos, a simples mutações nas regiões de seqüência do gene alfa que são muito importantes para a síntese de globina alfa. A maioria das não-deleções afetam especialmente a expressão de síntese do gene a2, agindo em regiões invariantes das bases nitrogenadas, formadas por guanina-timina, trocando-as por outra base, ou interferindo no processo de translação do RNAm maduro. São, portanto, mutações raras, restritas a determinadas regiões geográficas. Muitas das mutações de talassemias alfa por não-deleção ainda permanecem obscuras, sem que tenham sido caracterizadas. Talassemia Alfa Adquirida As talassemias alfa adquiridas
são de causas não-genéticas, que geralmente
se expressam como se fossem doenças de Hb H. A maioria dos
casos descritos aponta a associação com doenças
hematológicas, entre as quais se destacam a eritroleucemia,
doenças linfo e mieloproliferativas crônicas e agudas,
e anemia sideroblástica. Todos os casos confirmados e descritos
na literatura científica apresentam pontos comuns: corpos
de Hb H intra-eritrocitários, concentrações
de Hb H por eletroforese variável entre 10 e 60% e redução
da síntese de globina alfa. Os principais mutantes de globina beta: Hb S, Hb C e Hb E, podem ser significativamente afetados pela coexistência da talassemia alfa. Observa-se nesses casos que a concentração da hemoglobina variante, por exemplo, a Hb S, quando em heterozigose (Hb AS/Tal. alfa – figura 7.9), decresce proporcionalmente à quantidade de genes alfa afetados (tabela 7.2). Esse mesmo processo ocorre na Hb C (Hb AC) e Hb E (Hb AE). O efeito da talassemia alfa associada a essas hemoglobinas variantes se deve ao fato das cadeias beta (bA) estarem carregadas mais negativamente e, por isso, dimerizam-se com maior eficiência do que aquelas carregadas mais positivamente pela mutação sofrida: bS (Glu- ® Val0), Hb C (Glu-® Lis+) e Hb E (Glu- ® Lis+). Da mesma forma, essa variação que ocorre na formação de dímeros (aA bS) também difere em relação ao tipo de mutação que originou a talassemia alfa. Indivíduos homozigotos com deleção a3,7 têm maior concentração de Hb S (22%) do que aqueles com deleção -a4,2 (18%). A figura 7.10, mostra por eletroforese a interação entre Hb SS e doença de Hb H, com visível caracterização da Hb H fracionada difusamente por ser formada por tetrâmeros b4S. Esses tetrâmeros de globina bS são mais instáveis do que os tetrâmeros bA, cuja Hb H se apresenta mais compacta no fracionamento eletroforético (ver figura 7.1). Sob o ponto de vista da fisiopatologia em pacientes com Hb SS associada ao traço alfa talassêmico, os efeitos são minimizados. |
|||||||||||||||||||||||||||||||||||||||
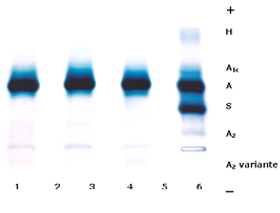
Figura 7.9 – Eletroforese de hemoglobinas em gel de agarose alcalina. Os casos 1, 2 e 3 pertencem a uma família com Hb A2 variante (1: mãe; 2: pai; 3: filho). O caso 4 é de um paciente com Hb AS associada a talassemia alfa caracterizada pela presença de Hb H com concentração de 8% (ASH). |
|||||||||||||||||||||||||||||||||||||||
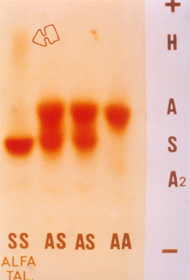
Figura 7.10 – Eletroforese de hemoglobinas em acetato de celulose, tampão alcalino. O primeiro caso à esquerda é de um paciente com anemia falciforme (Hb SS) associada à talassemia alfa caracterizada pela presença de Hb H difusa (b4S ). |
|||||||||||||||||||||||||||||||||||||||
Tabela 7.2 - Características hematológicas e laboratoriais da talassemia alfa/Hb AS. |
|||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||
Interação Talassemia Alfa/Talassemia Beta Os efeitos fisiopatológicos
das talassemias se devem ao desequilíbrio verificado entre
as globinas alfa e beta. A interação entre talassemias
alfa e beta diminui o grau do desequilíbrio alfa/beta, modificando
inclusive os quadros clínicos e hematológicos. O número
de genes alfa afetados, associados à talassemia beta homozigota,
pode produzir efeito benéfico. Os homozigotos para talassemia
b 0 (b
0/b 0 tal.) que não
são dependentes de transfusões são frequentemente
diagnosticados como portadores também de talassemia alfa,
e seus quadros clínicos se assemelham aos da talassemia intermédia. |
|||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||