Autor: Flavio Augusto Naoum |
||||||||||||
Quadro clínico da beta talassemia Na beta talassemia, o principal fenômeno responsável pelo desenvolvimento da anemia é a eritropoiese ineficaz com hemólise intra-medular dos precursores eritróides. Assim, nas apresentações mais leves da doença, a eritropoiese ineficaz é de cerca de 25%, enquanto que nos casos mais graves pode chegar até 90%. Isso justifica a discrepância comumente observada entre o aumento relativamente modesto do número de reticulócitos na vigência de quadros anêmicos muito intensos associados à hiperplasia acentuada da medula óssea.A beta talassemia apresenta basicamente três formas clínicas claramente distintas: 1) Talassemia menor ou minor (Traço talassêmico) Essa é a apresentação observada nos indivíduos heterozigotos para a mutação da beta talassemia, que em geral não causa complicações clínicas ou sintomas evidentes. No entanto, alguns indivíduos podem apresentar sinais de cansaço ou fraqueza quando submetidos a esforço prolongado ou atividade física. A ausência de sintomas ou sua menor intensidade nesses pacientes se deve ao fato de que a medula óssea consegue compensar a anemia causada pela discreta diminuição da produção de globinas beta nessa condição. Assim, o diagnóstico da talassemia menor é geralmente feito de forma acidental, a partir da investigação de um hemograma alterado realizado por motivos variados (“check-up”, exames ocupacionais, pré-operatórios, etc.) ou por meio de investigação familiar. No hemograma desses pacientes, os valores de hemoglobina estão geralmente normais ou discretamente diminuídos, embora haja microcitose, hipocromia e poiquilocitose evidentes no esfregaço sanguíneo. Com raras exceções, não há necessidade de tratamento específico para a talassemia menor. No entanto, a confirmação diagnóstica é muito importante, pois permite a orientação e o esclarecimento do paciente, evitando assim investigações e tratamentos desnecessários (como a suplementação equivocada de ferro), além de possibilitar o aconselhamento genético e eventual investigação familiar. |
||||||||||||
|
||||||||||||
2) Talassemia intermediária Talassemia intermédia é o termo utilizado para caracterizar indivíduos que apresentam quadro clínico intermediário entre as formas leves (talassemia menor) e graves (talassemia maior) da talassemia. Assim, nota-se grande variabilidade de sintomas e complicações nesses pacientes, desde quadros quase imperceptíveis até situações complicadas próximas às observadas na talassemia maior. Quanto à base genética dessa doença, observa-se na maioria dos casos homozigose para lesões parciais do gene da beta globina, heterozigose composta entre mutações da globina beta e outros defeitos nos genes alfa, delta ou gama, ou heterozigose com cadeias beta hiper-instáveis (talassemia dominante). Em termos gerais, os pacientes portadores dessa condição apresentam valores de hemoglobina entre 7 e 9g/dL, sensação de fraqueza e cansaço mais freqüente e necessidade de transfusões intermitentes (principalmente em caso de cirurgias e intensificação da anemia por outros motivos). O tratamento consiste de suplementação com ácido fólico, monitorização cuidadosa da intensidade da anemia e possíveis complicações como a sobrecarga de ferro, osteopatia, déficit de crescimento, entre outras. Na maioria dos casos não é recomendável tratamento com transfusões regulares. |
||||||||||||
|
||||||||||||
3) Talassemia maior ou major Trata-se da forma mais grave de beta talassemia. Os pacientes com talassemia maior geralmente apresentam lesões graves nos dois genes da globina beta, que podem resultar de herança em heterozigotose composta para duas mutações diferentes ou homozigose para a mesma mutação. A gravidade da lesão genética repercute no quadro clínico, causando anemia muito acentuada e fazendo com que os indivíduos afetados dependam de transfusões de sangue regulares para sobreviverem. Os primeiros sintomas costumam surgir após o sexto mês de vida, quando a quantidade de hemoglobina fetal diminui substancialmente e a síntese de hemoglobina A1 não ocorre devido à falta de produção de cadeias beta. Desse modo, é recomendável que as crianças afetadas sejam acompanhadas desde os primeiros meses de vida. Repercussões da anemia crônica Nos pacientes que não recebem tratamento transfusional adequado, a anemia intensa e persistente desde a infância pode acarretar complicações importantes como: • Palidez de pele e mucosas, além de icterícia. • Retardo no crescimento. • Estado hipermetabólico resultando em massa muscular deficiente, redução da gordura corporal, febre recorrente, falta de apetite e letargia. • Cardiopatia decorrente da intensificação do trabalho cardíaco para compensar o estado anêmico crônico. • Hepatoesplenomegalia (aumento do volume do fígado e do baço) persistente (Figura 1). A esplenomegalia progressiva pode precipitar os seguintes eventos: a) expansão do volume plasmático devido ao desvio de sangue para dentro do baço e da medula óssea hiperplasiada, causando hemodiluição e piorando a anemia; b) redução da sobrevida e seqüestro dos eritrócitos e também de outras células do sangue (hiperesplenismo); c) aumento da necessidade transfusional e, por conseqüência, da sobrecarga de ferro. • Hiperplasia de medula óssea acarretando mal-formações e deformidades ósseas como, por exemplo, o aumento do osso maxilar com exposição dos dentes da arcada superior e a proeminência malar causando o “achatamento” do nariz. Além disso, o aspecto radiológico do crânio, ossos longos e mãos, demonstra que a hiperplasia medular no interior dos ossos causa também o achatamento da camada cortical dos mesmos, tornando-os mais frágeis e susceptíveis a lesões e fraturas (Figuras 2 e 3). • Aumento da absorção de ferro pelo trato gastro-intestinal, por meio de um mecanismo compensatório equivocado deflagrado pela hiperplasia medular e pela anemia. |
||||||||||||
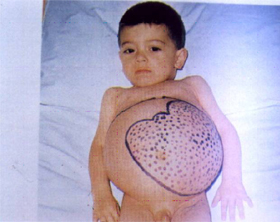
Figura 1: Aumento do volume abdominal causado pela esplenomegalia em criança portadora de talassemia maior. |
||||||||||||
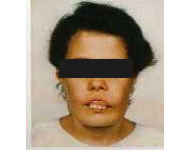
Figura 2: Deformidade óssea facial (hipertrofia do osso maxilar) em paciente com talassemia maior. |
||||||||||||
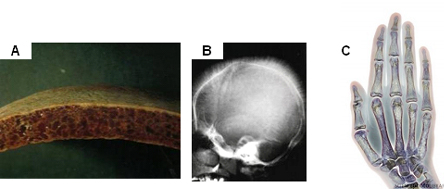 Figura 3: Adelgaçamento da cortical óssea
no crânio vista na macroscopia do osso (3A) e na radiografia
(aspecto de “terminações em cabelo”) (3B).
A figura 3C mostra desmineralização óssea (áreas
claras) vista na radiografia colorida da mão de paciente com
talassemia maior.
Figura 3: Adelgaçamento da cortical óssea
no crânio vista na macroscopia do osso (3A) e na radiografia
(aspecto de “terminações em cabelo”) (3B).
A figura 3C mostra desmineralização óssea (áreas
claras) vista na radiografia colorida da mão de paciente com
talassemia maior. |
||||||||||||
Repercussões da sobrecarga de ferro A necessidade de transfusões de sangue regulares desde a infância faz com que pacientes com talassemia maior desenvolvam inevitavelmente um quadro de sobrecarga de ferro no organismo que, juntamente com a anemia, torna-se também um dos principais riscos à saúde desses indivíduos. O ferro é um elemento importante que participa de várias reações químicas dentro e fora das células do organismo. A participação do ferro no metabolismo do organismo é biologicamente protegida pela ausência de via de excreção específica, o que facilita o acúmulo deste elemento quando há aumento de sua absorção ou introdução por outras vias (p.ex. transfusão de sangue). O excesso de ferro no organismo é prejudicial e potencialmente tóxico para diversos órgãos, e merece a adoção de medidas preventivas ou terapêuticas em pacientes de risco. Em condições normais, a quantidade de ferro presente no corpo humano gira em torno de 40 a 50mg por Kg de peso, sendo que a maior parte deste elemento (30mg/Kg) está incorporada à hemoglobina. Pacientes com talassemia recebendo transfusões regularmente acumulam cerca de 0,3 a 0,5mg de ferro por Kg por dia. Quando a quantidade de ferro acumulada atinge 12 a 24g, as complicações clínicas começam a aparecer. Se não for tratada, a sobrecarga de ferro geralmente leva os pacientes ao óbito na segunda ou terceira década de vida, principalmente por conta de complicações cardíacas. As principais complicações associadas à sobrecarga de ferro em diferentes tecidos e órgãos do corpo são: • Cardíacas: arritmias e insuficiência cardíaca (Figura 4). • Hipogonadismo hipogonadotrófico por lesão do hipotálamo e glândula pituitária, podendo resultar em baixa estatura, retardo ou ausência de puberdade, e infertilidade. • Outros problemas endócrinos: intolerância a glicose, diabetes mellitus, hipotiroidismo e hipoparatiroidismo. • Disfunção hepática: cirrose ou fibrose hepática (Figura 4). • Propensão à infecção (p.ex. Yersinia). A quantificação da sobrecarga
de ferro por meio de exames específicos é extremamente
importante para a tomada de decisões relacionadas ao tratamento
de quelação (retirada do ferro em excesso do organismo).
Os principais métodos utilizados para este fim são: |
||||||||||||
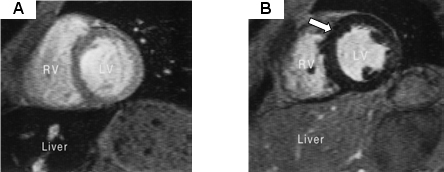 Figura 4: Ressonância magnética com
análise por meio da técnica do T2* (áreas escuras
denotam acúmulo de ferro). A) Acúmulo de ferro no fígado
(seta); acima do fígado, vê-se que o coração
não tem acúmulo significativo de ferro. B) Acúmulo
de ferro no coração (seta); fígado sem acúmulo
significativo. (Fonte: Anderson et al. Eur Heart J 2001;22:2171-9).
Figura 4: Ressonância magnética com
análise por meio da técnica do T2* (áreas escuras
denotam acúmulo de ferro). A) Acúmulo de ferro no fígado
(seta); acima do fígado, vê-se que o coração
não tem acúmulo significativo de ferro. B) Acúmulo
de ferro no coração (seta); fígado sem acúmulo
significativo. (Fonte: Anderson et al. Eur Heart J 2001;22:2171-9). |
||||||||||||
|
||||||||||||
O tratamento da talassemia O tratamento “standard” dos pacientes portadores de talassemia maior consiste basicamente em transfusões de sangue regulares a cada 3 ou 4 semanas, associadas ao uso dos quelantes de ferro (medicações que retiram o excesso de ferro acumulado por conta das transfusões de sangue). Há algumas décadas, a maioria dos pacientes com talassemia maior não sobrevivia além dos 20 ou 30 anos de idade. Com o modelo terapêutico atual, adotado na maioria dos centros especializados, é possível aumentar consideravelmente a expectativa de vida em boa parte dos pacientes, inclusive para próximo do normal. A melhora na expectativa de vida dos pacientes trouxe novos desafios terapêuticos que até então não haviam sido considerados como, por exemplo, a avaliação da fertilidade em pacientes adultos que planejam ter filhos ou mulheres que passarão por gestação, bem como a presença de outras comorbidades típicas de idade avançada, como hipertensão arterial, diabetes, DPOC, câncer, etc. A comunicação entre os pacientes e familiares, bem como entre os profissionais dos diferentes níveis de complexidade que atendem os pacientes é fundamental para que o tratamento proposto ocorra de forma eficaz. Centros de tratamento O acompanhamento e tratamento de pacientes portadores das formas graves de talassemia devem ser feitos preferencialmente em centros que apresentam as seguintes características: • Médicos hematologistas com especialização ou experiência em hemoglobinopatias. Profissionais da saúde (enfermeiros, fisioterapeutas, psicólogos e assistentes sociais) com experiência ou capacitação em hemoglobinopatias. • Infra-estrututra para atendimento ambulatorial, emergência 24h e internação de pacientes. Laboratório de análises clínicas e recursos para realizar exames de imagem complexos. Banco de sangue próprio ou conveniado e hospital-dia para acomodar pacientes que recebem transfusões regulares e programadas. • Facilidade para encaminhar pacientes para avaliação por outras especialidades. • Centro de apoio próprio ou conveniado para oferecer suporte psicológico, aconselhamento genético, orientações e reuniões de grupos de pacientes e familiares. Transfusões de sangue Transfusões de sangue regulares desde a infância consistem na melhor e principal forma de tratamento para pacientes portadores de talassemia maior (Figura 5). Os principais objetivos do tratamento transfusional são: corrigir a anemia, permitir o crescimento, desenvolvimento e realização de atividades normais, prevenir o aumento do baço e inibir a hiperplasia eritróide na medula óssea. Por definição, pacientes com talassemia intermédia ou doença da hemoglobina H não necessitam de receber transfusões regulares. Entretanto, transfusões ocasionais podem ocorrer por motivos de doenças, gestação ou cirurgias, entre outras. Assim, as medidas terapêuticas que serão descriminadas a seguir referem-se principalmente ao tratamento de pacientes com talassemia maior. |
||||||||||||

Figura 5: Criança com talassemia maior recebendo transfusão de sangue. (Fonte: www.thalassaemia.org) |
||||||||||||
Início das transfusões Antes do início do tratamento transfusional, é desejável que se faça a fenotipagem extensa de grupos sanguíneos, uma vez que esta pode ser dificultada pela formação alo-anticorpos após múltiplas transfusões. Apesar da possível interferência do genótipo na gravidade da evolução da talassemia maior, a decisão de quando se iniciar o tratamento transfusional permanece eminentemente clínica. Assim, geralmente considera-se iniciar o programa de transfusões regulares nas crianças com talassemia maior quando o valor da hemoglobina é menor que 7g/dL com presença de fadiga importante, alimentação deficiente, retardo ou regressão no desenvolvimento, retardo no crescimento. Outros fatores a serem considerados nessa decisão são esplenomegalia progressiva e evidência de expansão ou deformação óssea. É importante excluir causas reversíveis de anemia associadas a talassemia, como ferropenia e quadros infecciosos concomitantes. Se possível, o tratamento transfusional deve ser iniciado até a idade de 3 anos, uma vez a partir daí o risco de desenvolver múltiplos anticorpos aumenta, tornando difícil a seleção de unidades compatíveis. Quanto e quando transfundir Geralmente costuma-se transfundir o paciente para manter o valor de hemoglobina ente 9,5 e 10g/dL. Nessas condições, é possível permitir o crescimento e desenvolvimento adequado das crianças afetadas, além de inibir a hiperplasia da medula óssea e minimizar o acúmulo de ferro. O intervalo entre as transfusões geralmente varia de 2 a 4 semanas e a quantidade de concentrados de hemácias varia de 1 a 3 unidades por vez (ou 10 a 15ml por Kg de peso). O ideal é que os pacientes pré-estabeleçam os dias das suas transfusões de acordo com a disponibilidade do serviço de hemoterapia e também de suas próprias ocupações. Normalmente, os pacientes realizam os exames pré-transfusionais alguns dias antes das transfusões. No dia das transfusões, é preferível que a canulação venosa seja feita por profissionais experientes, uma vez que várias tentativas de punção sem sucesso tendem a tornar o procedimento cada vez mais difícil nas transfusões subseqüentes, além de aumentar a ansiedade do paciente. As unidades transfundidas devem ser leucodepletadas e compatíveis em relação aos grupos sanguíneos ABO, Rh (D, C, c, E,e) e Kell. A seleção de unidades com maior volume de glóbulos vermelhos e menor tempo de coleta (menos de duas semanas, se possível) pode ajudar a diminuir a freqüência das transfusões no paciente. Complicações do tratamento transfusional A principal complicação relacionada ao regime transfusional crônico é a sobrecarga de ferro. Outras complicações importantes associadas às transfusões de concentrados de hemácias são: • Reações transfusionais agudas (febril hemolítica e não-hemolítica, e urticariforme, entre outras) • Transmissão de infecções (p.ex. hepatites virais e HIV) • Alo-imunização É importante destacar que embora exista o risco de transmissão de infecções por meio de transfusões, o mesmo encontra-se muito reduzido em virtude das políticas rigorosas de triagem clínica e sorológica dos doadores adotadas em praticamente todos os bancos de sangue e hemocentros do Brasil. Da mesma forma, boa parte das reações transfusionais agudas são minimizadas pela utilização do filtro de leucócitos nas unidades transfundidas, assim como a incidência de alo-imunização diminui com a transfusão de unidades fenotipadas para os grupos sanguíneos ABO, Rh e Kell. Sobrecarga de ferro Geralmente a terapia quelante tem seu início em crianças por volta dos 3 anos de idade, após terem recebido cerca de 10 a 12 transfusões ou quando o valor da ferritina encontra-se persistentemente acima de 1000µg/L. Atualmente, os quelantes disponíveis no mercado são: 1) Desferrioxamina (Desferal®): é o quelante mais antigo e tradicional, utilizado como monoterapia por décadas. Devido ao grande tamanho da molécula desta droga, a absorção intestinal torna-se impossibilitada. Assim, seu principal inconveniente é a via de administração, necessariamente subcutânea ou endovenosa, e o tempo prolongado de infusão (Figura 6). A dose inicial preconizada é de 25-30mg/Kg por infusão subcutânea de 8 a 12 horas, administrada cinco a seis vezes por semana. Embora eficaz, o tratamento com desferrioxamina é geralmente prejudicado pela falta de adesão ao protocolo por parte do paciente. Os principais efeitos colaterais desta medicação são: retardo no crescimento, lesões ósseas e perda da acuidade auditiva. 2) Deferiprone (L1): foi o primeiro quelante oral a ser utilizado em grande escala nos pacientes (preferencialmente acima dos 6 anos de idade). Mostrou-se mais eficaz na remoção do ferro cardíaco em comparação com os outros quelantes. Pode ser utilizado como monoterapia ou em combinação com a desferrioxamina. A dose inicial preconizada é de 75mg/Kg/dia por via oral dividida em três tomadas (de 8 em 8 horas). Os principais efeitos colaterais são: agranulocitose (0,5-1%), artropatia (4-50%), náuseas, elevação intermitente de transaminases. Na terapia combinada com desferrioxamina, o deferiprone deve ser tomado diariamente e a dose e freqüência da desferrioxamina ajustada em função da sobrecarga de ferro. 3) Deferasirox (Exjade®): novo quelante oral com resultados promissores e posologia cômoda, embora com pouco tempo de uso. Mostrou-se muito eficaz na remoção do ferro hepático em comparação com os demais quelantes. Pode ser usado após os 2 anos de idade. A dose inicial preconizada é de 20mg/Kg/dia por via oral uma vez ao dia. Os principais efeitos colaterais são sintomas gastro-intestinais (náuseas, vômitos e diarréia), rash cutâneo, nefrotoxicidade (elevação da creatinina sérica). |
||||||||||||
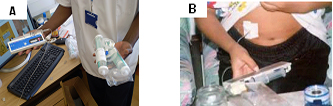 Figura 6: Tipos de bomba (A) para infusão da desferrioxamina e aplicação subcutânea da droga (B). |
||||||||||||
Transplante de medula óssea Até o momento, já foram transplantados mais de 1600 pacientes com talassemia maior. Na grande maioria dos casos este tratamento tem sido realizado em pacientes com menos de 17 anos de idade, sendo que a idade ideal para a realização deste procedimento situa-se entre os 18 meses e 3 anos. Embora seja o único tipo de tratamento capaz de curar pacientes com talassemia maior, a ampla utilização do transplante de medula óssea (TMO) enfrenta os seguintes obstáculos: • Disponibilidade de doador: apenas 30% das crianças com talassemia maior apresenta um doador aparentado (irmão ou irmã) HLA-compatível. • Estado geral e intensidade das complicações decorrentes da talassemia no momento da indicação do TMO. • Toxicidade e complicações associadas ao TMO: infecções, doença do enxerto contra hospedeiro (aguda e crônica), tempo de aplasia prolongado, sangramentos, disfunções endócrinas, risco de neoplasia secundária, etc. • Mortalidade associada ao procedimento de 7 a 15% nos centros mais experientes, incluindo pacientes de alto e baixo risco. • Melhora do tratamento de suporte: tratamento transfusional mais seguro e eficaz, novos quelantes com posologia cômoda, aumento considerável da sobrevida com tratamento conservador, etc. Os fatores de risco que sabidamente prejudicam o resultado do TMO na talassemia maior são: • Tratamento quelante irregular • Hepatomegalia • Fibrose hepática (quantificada por meio de biópsia) Nos pacientes sem nenhum fator de risco a sobrevida global, sobrevida livre de talassemia e a mortalidade são, respectivamente, de 93%, 91% e 7%, e naqueles com os três fatores de risco presentes, de 79%, 58% e 10%. Embora a indicação de TMO na talassemia maior não seja rotineira, nem automática, a recomendação é que o assunto deva ser discutido com a família quando a criança afetada tem entre 12 e 18 meses de idade, independente da disponibilidade ou não de doador. Os detalhes referentes ao TMO devem ser esclarecidos por uma equipe especializada neste tipo de tratamento e a decisão tomada em conjunto entre médicos, paciente e seus familiares (principalmente no caso de crianças). Momentos cruciais do acompanhamento dos talassêmicos Pacientes portadores de hemoglobinopatias podem e devem ser acompanhados também por pediatras, clínicos gerais, ginecologistas e médicos de família, que estão habilitados a avaliar e resolver boa parte dos problemas de saúde apresentados pelos pacientes, especialmente os de origem não hematológica. Entretanto, há determinados momentos que demandam uma avaliação mais especializada. As situações descritas abaixo devem ser abordadas preferencialmente em centros de referência para tratamento de pacientes portadores de hemoglobinopatias: • Confirmação do diagnóstico • Início do tratamento transfusional • Início da terapia quelante • Decisões quanto ao tipo e dose do tratamento quelante • Problemas de adesão ao tratamento quelante • Revisão anual clínica e laboratorial do paciente com talassemia • Discussões acerca de decisões potencialmente complexas: indicação de esplenectomia e cirurgias de grande porte; ação diante de problemas e disfunções endócrinas, ortopédicas, cardíacas, hepáticas e de fertilidade; indicação de transplante de medula óssea; problemas psico-sociais complexos. Bibliografia 1. Naoum PC. Hemoglobinopatias e talassemias. 1ª ed. São Paulo: Sarvier; 1997. 2. Steinberg et al. Disorders of hemoglobin. 1ª ed. Cambridge University Press; 2001. 3. Rodgers GP. Sickle cell disease and thalassaemia. Cambridge: Baillière Tindal; 1998. 4. Yardumian et al. Standards for the clinical care of children and adults with thalassaemia in the UK. United Kingdom Thalassaemia Society; 2005. 5. Hoffbrand AV, Pettit JE. Clinical Haematology. 2nd ed. Sandoz, 1994. 6. Anderson et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J 2001;22:2171-9. 7. Bhatia M, Walters MC. Hematopoietic cell transplantation for thalassemia and sickle cell disease. Bone Marrow Transplantation 2008;41:109-17. |
||||||||||||
 |
||||||||||||
|
||||||||||||