Flávio Augusto Naoum
A interação entre Hb S e talassemia beta foi descrita pela primeira vez por Ida Bianco e Ezio Silvestroni em 1954. Esses pesquisadores observaram que alguns dos seus pacientes apresentavam anemia de graus moderado a grave, com a presença concomitante de células falciformes e microcíticas, e por essa razão a anemia foi denominada de micro-drepanocitose. Eletroforeticamente a interação Hb S/Talassemia Beta apresenta dois padrões genotípicos, um com as hemoglobinas SF (Hb SF) e outro com Hb SFA; em ambos os casos a concentração da Hb S é sempre maior que as outras hemoglobinas.
A ocorrência da interação Hb S/Talassemia Beta em nosso país se deve ao alto grau de miscigenação entre as etnias que participaram da formação do povo brasileiro. Embora o gene da globina bS seja muito prevalente entre populações do continente africano, outros povos também apresentam altas prevalências notadamente na Arábia Saudita, Emirados Árabes, Caribe, sul da Itália, e em negros das Américas do Norte e do Sul. Por outro lado, o gene da globina b anormal que induz a falha na síntese da globina beta – ou gene b talassêmico – apresenta expressiva prevalência entre os povos de países banhados pelo mar Mediterrâneo, com destaques para Itália, Turquia, Grécia, Chipre, Itália, Líbano e países do norte da África. Ao longo do tempo ocorreu a expansão do gene b talassêmico para Portugal, Espanha, leste europeu e Ásia. No Brasil a prevalência da Hb S/Talassemia Beta é de um caso para 25 mil pessoas, e conforme dados obtidos junto à Organização Mundial da Saúde estima-se que nascem anualmente no Brasil cerca de 600 crianças com esse genótipo.
A Hb S/Talassemia Beta pode ser diferenciada laboratorialmente por meio de eletroforeses alcalina e ácida em dois genótipos: SF e SFA. No genótipo SF há completa ausência da Hb A, com concentrações variáveis de Hb A2 entre 2 e 6% conforme mostra o caso n.º 3 da figura 6.41 e o caso n.º 3 da figura 6.42 – especialmente neste paciente há discreta concentração de Hb A proveniente de transfusão de concentrado de hemácias.
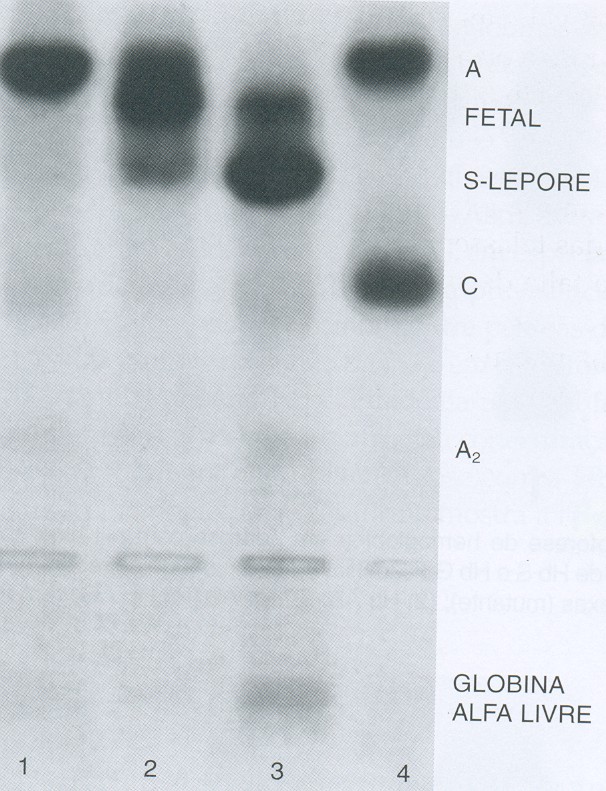
Figura 6.41: Eletroforese de hemoglobinas em agarose alcalina mostrando em (1) Hb Lepore heterozigota com concentração de 5%; (2) Hb Lepore homozigota, com Hb Fetal ± 70%, Hb Lepore ± 15%, Hb A (transfundida) ± 15%, e traços de Hb A2; (3) Hb SF de um paciente com Hb S/b0 talassemia, com globinas alfa livre; (4) Hb AC.
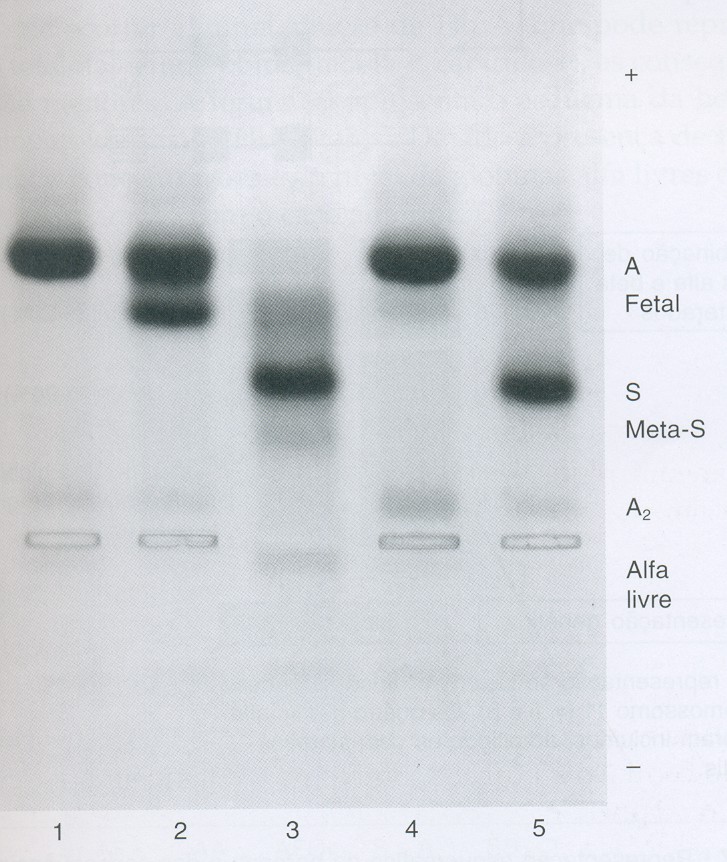
Figura 6.42 – Eletroforese alcalina de hemoglobinas em gel de agarose. (1) Hb AA – padrão normal; (2) Hb AF na talassemia beta maior após transfusão de concentrado de hemácias; (3) Hb S/Tal. b0 ou Hb SF com excesso de metaemoglobina S e globinas alfa livres; (4) Pai do caso 3 – talassemia beta menor com Hb A2 e Hb Fetal aumentadas; (5) Mãe do caso 3 com Hb AS.
Esse genótipo se deve à total deficiência de síntese de globina beta normal ou bA, sendo por isso denominado por talassemia b0 que associado a Hb S se torna Hb S /Tal.b0. Na talassemia b0 há elevada expressão do gene gama que induz o aumento da síntese da Hb Fetal quando há interação com a Hb S. A situação mais freqüente de herança desse genótipo ocorre quando um dos pais é portador heterozigoto de talassemia beta menor, ou Tal. b0 heterozigoto (figura 6.43), e o outro é portador de Hb AS, conforme mostra a figura 6.44.
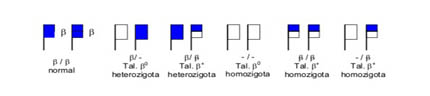
Figura 6.43: Representação esquemática de genes beta normal e nas talassemias b0 e b+, nos pares de cromossomo 11.
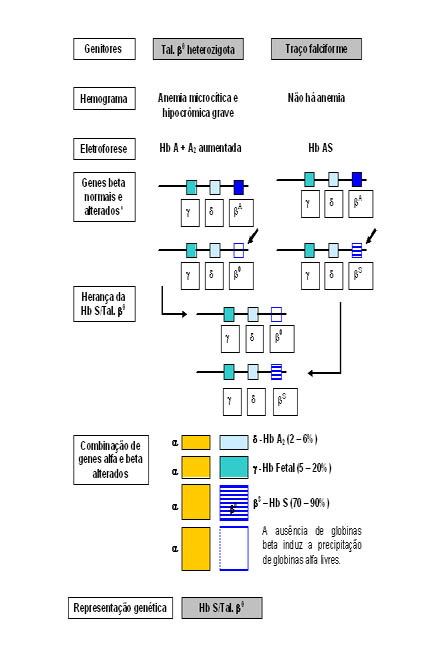
* Nesta representação só foram incluídos os genes do cromossomo 11 (g, d e b). Os quatro genes alfa não foram incluídos, admitindo-os com sínteses normais.
Figura 6.44 – Representação esquemática da herança e das conseqüências fisiopatológicas da Hb S/Tal. b0.
Pela representação esquemática dessa figura é possível entender a ausência eletroforética da Hb A, e as concentrações elevadas das Hb S e Hb Fetal. Entretanto um fato relevante mostrado no esquema se refere às globinas alfa "livres" que por não terem globinas bA para se combinarem, precipitam nos eritrócitos e formam corpos de Heinz. Os corpos de Heinz são retirados por macrófagos do sistema retículo endotelial, causando lesões nos eritrócitos – principal causa da formação de micrócitos esquisócitos e, consequentemente da microcitose. As globinas alfa "livres" são facilmente identificadas em eletroforese alcalina de hemoglobina em agarose ou acetato de celulose, que após coloração com negro de amido são visíveis atrás do ponto de aplicação das amostras (caso 3 da figura 6.42). Destaque importante dessa figura se deve à presença de meta Hb S induzida pelo alto grau de oxidação da Hb S (ver figura 6.45) e também, em parte causada pela oxidação das globinas alfa "livres". Como já foi exposto anteriormente, o alto grau de oxidação da hemoglobina inviabiliza a sua integridade molecular, tornando-a instável estruturalmente – principal causa da precipitação de globinas bS sob forma de corpos de Heinz.
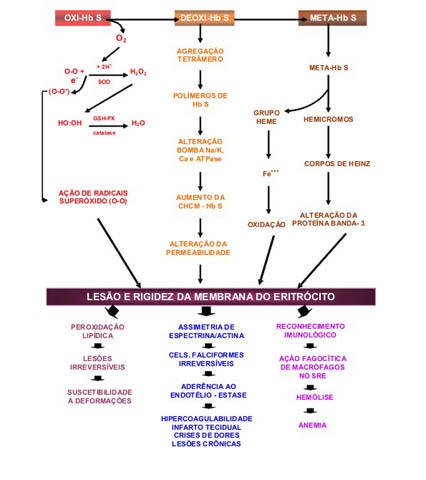
Figura 6.45 – Conseqüências celular e fisiopatológica da desoxigenação da Hb S.
O genótipo SFA se deve à associação entre a talassemia b+ - caso em que o gene da globina bA está parcialmente afetado para sintetizar Hb A – e Hb S, configurando a representação genética de Hb S/Tal.b+. Nesse caso a forma mais comum da expressão dessas alterações se caracteriza pela maior concentração da Hb S em relação à Hb A (figura 6.46).
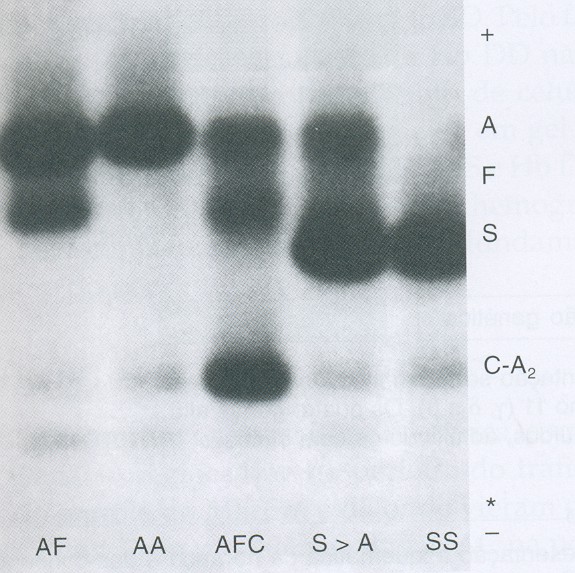
Figura 6.46 – Eletroforese alcalina de hemoglobinas em acetato de celulose. Da esquerda para a direita: AF (talassemia beta maior após transfusão de concentrado de hemácias); AA (padrão normal); AFC (recém-nascido com Hb AC e Hb Fetal elevada devido à idade); S>A (Hb S/Tal. b+, com Hb S mais concentrada que a Hb A; SS (Anemia falciforme).
Eventualmente a Hb Fetal pode estar elevada. Pelo fato de ocorrer alguma síntese de Hb A, que pode representar entre 10 e 40% do total da hemoglobina, as conseqüências fisiopatológicas são menores. A figura 6.47 apresenta o esquema da herança e conseqüências fisiopatológicas da Hb S/Tal. b+. Devido a presença de Hb A – mesmo que em baixas concentrações – o nível de globinas alfa livres é nitidamente inferior em comparação com o caso anterior.
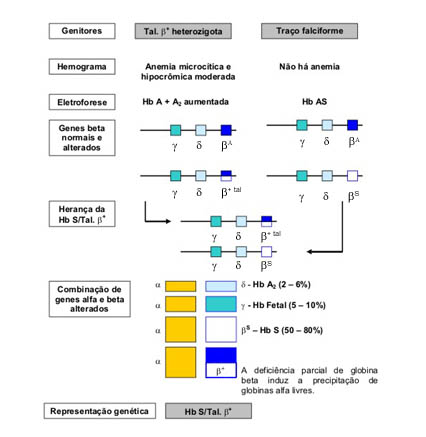
* Nesta representação só foram incluídos os genes do cromossomo 11 (g, d e b). Os quatro genes alfa não foram incluídos, admitindo-os com sínteses normais.
Figura 6.47 – Representação esquemática da herança e das conseqüências fisiopatológicas da Hb S/Tal. b+.
É fundamental a realização dos exames eletroforéticos dos pais de portadores de Hb S/Tal. b, onde um deles deve apresentar Hb S (Hb AS, Hb SC, Hb SS ou Hb S/Tal. b) e o outro talassemia beta (Tal. b menor, Hb S/Tal. b ou Hb C/Tal. b). Entretanto os genótipos mais comuns dos pais são: Hb AS x Hb Ab (Tal. b menor). Essa avaliação é necessária porque muitas vezes é transfundido sangue com Hb AA para pessoas com anemia falciforme (Hb SS), e nesses casos o resultado eletroforético pós-transfusional é similar ao da Hb S/Tal. b+, com a concentração da Hb S maior que a Hb A.
Além do hemograma, com destaque para os índices VCM, HCM e resultados das análises por eletroforeses alcalina e ácida, é necessário que se faça a dosagem de Hb Fetal para o correto estabelecimento do diagnóstico laboratorial da Hb S/Talassemia Beta. A dosagem de metaemoglobina, contagem de reticulócitos, e pesquisa intraeritrocitária de corpos de Heinz constituem excelentes parâmetros para avaliar as conseqüências fisiopatológicas da doença.
