Flávio Augusto Naoum
A anemia falciforme é o estado de homozigose da Hb S representado por Hb SS. Ao contrário dos portadores do traço falciforme (Hb AS), a história de pacientes com anemia falciforme revela, quase invariavelmente, por repetidos episódios dolorosos, geralmente do tipo vasculoclusivo, com ou sem efeitos mielodepressivos (deficiência temporária da medula óssea em produzir eritrócitos). Os sinais e sintomas são especialmente causados por anemia crônica, períodos de agravamento de anemia, dores nas juntas e nas extremidades de mãos e pés, dores abdominais, necrose asséptica da medula óssea, acidentes cerebrovasculares, infartos pulmonares, entre outros.
Os recém-nascidos com anemia falciforme geralmente não apresentam os problemas causados por essa hemoglobinopatia devido à alta concentração de Hb Fetal presente nos eritrócitos, durante os dois primeiros meses de vida. Entretanto, a gradual substituição da Hb Fetal (a2 g2 ) pela Hb S (a2 b2S), motivada pela redução genética da síntese de globinas g ao mesmo tempo em que ocorre a indução genética da síntese de globinas bS, faz com que a doença se expresse a partir do quarto mês de vida.
Os exames laboratoriais indicam anemia grave, com hemoglobina variável entre 5 e 9 g/dl, que associados à acentuada queda de hematócrito e contagem de eritrócitos resultam em índices normocíticos e normocrômicos, apesar da evidente aniso-poiquilocitose com presença de células falciformes. A leucocitose é moderada (12.000 a 17.000/mm3), freqüentemente com desvio à esquerda com neutrófilos bastonetes. As plaquetas podem estar discretamente elevadas. O exame da medula óssea apresenta hiperplasia normoblástica e áreas de hematopoiese podem ser encontradas em regiões geralmente ocupadas por tecido adiposo (ou medula amarela). Os exames bioquímicos do sangue revelam alterações em vários parâmetros, conforme mostra a tabela 6.9.
Tabela 6.9 – Parâmetros bioquímicos em 74 pacientes com anemia falciforme do Instituto Estadual de Hematologia do Rio de Janeiro – HEMORIO.
Parâmetros |
Valores obtidos |
Média |
Ferritina (mg %) |
16,4 – 1000,0 |
223,07 |
Bilirrubina total (mg %) |
0,4 – 8,8 |
2,65 |
Bilirrubina direta (mg %) |
0,1 – 2,6 |
0,62 |
Bilirrubina indireta (mg %) |
0,3 – 8,1 |
2,04 |
AST (UI/L) |
14,0 – 129,0 |
45,37 |
ALT (UI/L) |
6,0 – 85,0 |
25,14 |
Creatinina (mg %) |
0,2 – 1,0 |
0,56 |
AST – Aspartato amino transferase (ou transaminase oxaloacética)
ALT – Alanina amino transferase (ou transaminase pirúvica)
Fonte: Marcos Kneip Fleury, 2000.
A eletroforese de hemoglobina apresenta concentrações de Hb S variáveis entre 90 e 100%, Hb Fetal: 2 a 10%, e Hb A2: 2 a 4%. A Hb A está sempre ausente em pacientes com anemia falciforme não transfundidos, conforme mostra a figura 6.39.
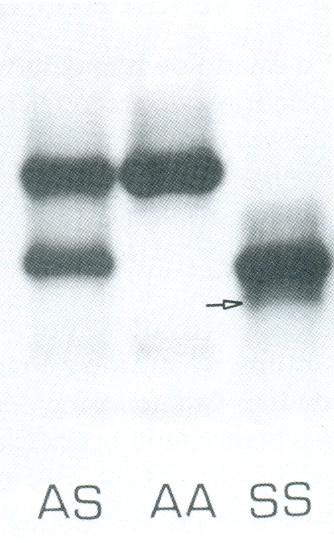
Figura 6.39 – Eletroforese alcalina de hemoglobina em acetato de celulose. Da esquerda para a direita: Hb AS, HB AA e Hb SS com 5% de Hb Fetal. A seta indica fração de meta-Hb S em paciente com anemia falciforme associada à deficiência de G-6-PD.
A Hb Fetal na anemia falciforme – Na anemia falciforme a concentração de Hb Fetal é geralmente variável entre 2 e 10%, mas pode ser maior que 10%, e em alguns casos pode ultrapassar 40%. Pesquisas realizadas em pacientes com anemia falciforme dos sexos masculino e feminino, revelaram que o nível médio e Hb Fetal é maior entre as mulheres. A concentração de Hb Fetal é determinada por fatores relacionados e não-relacionados com o agrupamento de genes da globina beta, fato evidenciado entre os diferentes haplótipos de Hb S obtidos de estudos realizados em pessoas com anemia falciforme. A análise dessa relação revela que a gravidade clínica da anemia falciforme é menor quando o nível de Hb Fetal está mais elevado. A relação entre os diferentes haplótipos de Hb S com a concentração de Hb Fetal parece estar relacionada com a persistência hereditária de Hb Fetal (PHHF) dos genes específicos para síntese da globina gama. No cromossomo 11 há dois genes gama que expressam globinas gamas diferentes – um deles com a síntese de alanina e outro com a síntese de glicina – e por isso denominados por gene gA e gene gG. Nas pequenas concentrações de Hb Fetal presente em adultos com hemoglobinas normais, a relação gG / gA, tende a um valor próximo de 2:3. Nas colônias de células eritróides obtidas de sangue fetal a Hb Fetal tem maior proporção de gG, enquanto que em colônias com maior concentração de Hb A, o predomínio é de gA. A porcentagem de células F, ou seja, eritrócitos com Hb Fetal, está aumentada na anemia falciforme. Estudos realizados com amostras de sangue de pacientes com anemia falciforme, em comparação com Hb AA, revelaram que cerca de 55% dos eritrócitos falciformes continham Hb Fetal, enquanto que em portadores de Hb AA o valor médio foi de 3%. Foi demonstrado também que fatores genéticos ligados ao cromossomo X interferem na síntese de Hb Fetal, fato que explicaria a maior concentração de Hb Fetal entre as mulheres.
Hb SS / Talassemia alfa (Hb SH) – A talassemia alfa é uma alteração da síntese de globina alfa que pode ter origem genética ou adquirida. A causa hereditária da talassemia alfa está bem definida e se deve a defeitos no processo de síntese de globinas alfa, por lesão molecular de um, dois, três ou quatro genes alfa. A causa adquirida tem sido relatada em pacientes com doenças linfo e mieloproliferativas, cuja prevalência de talassemia alfa chega atingir 60% entre esses doentes. Entretanto, pouco se sabe de que forma surge a talassemia alfa adquirida – se induzida por drogas quimioterápicas ou por alterações genéticas da célula tronco hematopoiética.
Entre pessoas negras, a talassemia alfa resulta geralmente da deleção de um ou dois genes alfa. A prevalência de talassemia alfa entre negros da América do Norte é de 30%, enquanto no Brasil é variável entre 20 e 25%. Assim, admite-se que a interação entre a anemia falciforme e talassemia alfa ocorra com alta frequência. Foi demonstrado em vários estudos descritos na literatura científica que a talassemia alfa representa um fator modulador dos parâmetros hematológicos, com elevação da contagem de eritrócitos e redução dos índices hematimétricos: VCM (volume corpuscular médio), HCM (hemoglobina corpuscular média), e CHCM (concentração de hemoglobina corpuscular média). A redução do número de moléculas de Hb S no eritrócito diminui a formação de polímeros das moléculas de desoxi-HbS, e consequentemente se forma menos eritrócitos falciformes irreversíveis. Dessa forma, a interação entre Hb S/talassemia alfa parece ser benéfica à anemia falciforme pois aumenta a sobrevida do eritrócito falcêmico, diminui o grau de hemólise, e consequentemente torna a anemia menos grave, conforme mostra a tabela 6.10.
Tabela 6.10 – Parâmetros hematológicos na interação entre anemia falciforme e talassemia alfa.
Genótipo |
Tipo de Hb |
Hb g/dl |
VCM |
Reticulócito |
a2 b2S / a2 b2S |
SS |
5 – 9 |
85 – 110 |
10 – 20 |
-a b2S / a2 b2S |
SH* |
7 – 10 |
80 – 95 |
5 – 10 |
-a b2S / -a b2S |
SH** |
9 – 11 |
70 – 80 |
5 – 10 |
-a b2S / - - b2S |
SH*** |
5 – 8 |
60 – 70 |
> 20 |
* Lesão de um gene alfa; Hb H: 0,5 a 2% na eletroforese alcalina.
** Lesão de dois genes alfa; Hb H: 2 a 8% na eletroforese alcalina.
*** Lesão de três genes alfa ou Hb S/doença de Hb H; Hb H: 15 a 25% na eletroforese alcalina.
Entretanto, na associação entre Hb S e doença de Hb H (lesão de três genes alfa) a anemia se torna grave. Nesses casos a concentração de Hb H associada à Hb S é variável entre 15 e 25% figura 6.40.
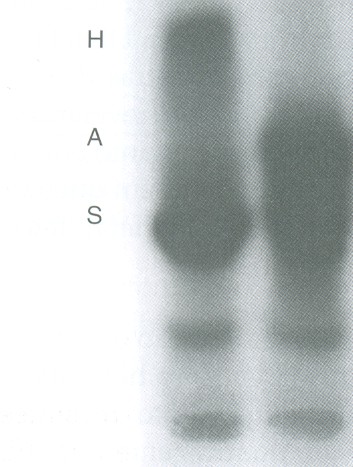
Figura 6.40 – Eletroforese alcalina de hemoglobina em acetato de celulose. À esquerda: Hb SH obtida de paciente com anemia falciforme associada a talassemia alfa (- a b2S / - - b2S), com concentração de Hb H de 23%. À direta: Hb AS. Para visualizar a Hb H é necessário aplicar grande quantidade de hemolisado na eletroforese, dificultando a separação de frações próximas como são os casos de Hb A, Fetal e S.
Anemia falciforme e deficiência de G-6-PD – A coexistência da Hb SS com deficiência de glicose-6-fosfato desidrogenase (G-6-PD) é um fato presumível uma vez que a prevalência da deficiência dessa enzima entre a população negra é por volta de 9 a 15%. Embora a literatura científica se refere de forma genérica que a associação entre Hb SS/deficiência de G-6-PD parece não ter influência no número de eritrócitos falciformes irreversíveis (ou células densa), ou no nível de concentração da hemoglobina total, bem como na condição clínica do paciente, é importante destacar que há, pelo menos, cinco classes diferentes de deficiência de G-6-PD (tabela 6.11).
Tabela 6.11 – Classificação da deficiência de G-6-PD em cinco classes.
Classes |
Características |
1 |
Variantes deficientes associadas à anemia hemolítica crônica. |
2 |
Variantes com deficiência acentuada da atividade da enzima G-6-PD, com menos de 10% de atividade normal. Ex.: variantes mediterrâneas. |
3 |
Variantes com deficiência moderada da atividade da enzima G-6-PD, com 10 a 60% da atividade normal. Ex.: variantes africanas. |
4 |
Variantes com deficiência discreta da atividade da enzima G-6-PD, com 60 a 95% da atividade normal. |
5 |
Variantes com atividade aumentada da enzima G-6-PD. |
A classe 1 é muito rara, entretanto as classes 2 e 3 são as mais importantes sob o ponto de vista clínico, pois causam crises de hemólises quando o portador é exposto a drogas oxidantes. No Brasil, a deficiência de G-6-PD mais comum é a da classe 3, com os tipos africanos prevalecendo em 87% dos casos descritos, seguida da classe 2 onde os tipos mediterrâneos atingem 13%.
Sob o ponto de vista fisiológico a associação da Hb SS com deficiência de G-6-PD é desastrosa à molécula de hemoglobina. A deficiência de G-6-PD é um fator indutor da desnaturação da hemoglobina, enquanto que a Hb S apresenta alto grau de oxidação espontânea. A associação desses dois defeitos num mesmo eritrócito certamente desencadeia com maior intensidade a desnaturação oxidativa da Hb S, especialmente se exposto a fatores indutores como são os casos de drogas e poluentes ambientais oxidativos. Diante desses fatos ocorre maior consumo das enzimas antioxidantes, alterando as sínteses e as suas concentrações. Assim, os mecanismos redutores da oxidação e das lesões celulares provocadas pelos radicais livres causam a diminuição das atividades de glutatião peroxidase e catalase, aumenta o nível de superóxido dismutase (SOD) e diminui a concentração de vitamina E no plasma e na membrana eritrocitária.
A avaliação citológica de corpos de Heinz nos pacientes com anemia falciforme é sempre um excelente marcador biológico dos processos oxidativos. Na associação entre Hb SS e deficiência de G-6-PD a precipitação intraeritrocitária de corpos de Heinz é muito mais expressiva. Outro indicador da deficiência de G-6-PD em pacientes com anemia falciforme é a elevada concentração de metaemoglobina, que pode ser quantificada bioquimicamente ou visualizada durante o processo eletroforético da Hb SS – notadamente após 48 horas da coleta do sangue (figura 6.39).
Investigação laboratorial – O diagnóstico laboratorial da anemia falciforme inclui o hemograma completo, as eletroforeses de hemoglobina em tampões alcalino e ácido, dosagens de Hb Fetal e metaemoglobina, contagem de reticulócitos e pesquisas intraeritrocitárias de Hb H e corpos de Heinz. O hemograma é fundamental para avaliar quantitativamente o grau de anemia, além de fornecer os índices hematimétricos de VCM e HCM que são importantes parâmetros para diferenciar os genótipos da anemia falciforme (SS e SF) e da talassemia beta (SF), conforme mostra a tabela 6.12.
Tabela 6.12 – Diferenciação dos principais genótipos de Hb S relacionados aos valores de hemoglobina (Hb g/dl), volume corpuscular médio (VCM), reticulócitos e Hb Fetal.
Doença |
Genótipo |
Hb (g/dl) |
VCM (fl) |
Reticulócitos (%) |
Hb Fetal (%) |
Anemia Falciforme |
bS / bS |
5 – 9 |
85 – 110 |
10 – 20 |
2 – 10 |
Hb S/tal a* |
bS / bS |
6 – 10 |
70 – 80 |
5 – 10 |
2 – 20 |
Hb SC |
bS / bC |
9 – 13 |
75 – 85 |
5 – 10 |
1 – 5 |
Hb S/tal b0 |
bS / b0 |
7 – 10 |
60 – 70 |
5 – 15 |
5 – 20 |
Hb S/tal b+** |
bS / b+ |
9 – 11 |
60 – 70 |
5 – 10 |
5 - 10 |
* - Deleção de dois genes alfa
** - Em eletroforese identificam-se as frações de Hb A, F e S; a Hb S tem concentração maior que a Hb A.
A eletroforese de hemoglobina em pH alcalino identifica a fração anormal com suspeita de ser a Hb S, pois nesta mesma região eletroforética se posiciona a Hb D. Assim, os genótipos SS, SD e DD apresentam com as mesmas características eletroforéticas. Para identificar cada um desses genótipos é necessário recorrer à eletroforese de hemoglobinas em gel de agarose com pH ácido, que separa com excelente resolução esses genótipos. É importante destacar ainda que a eletroforese alcalina, quer seja em gel de agarose ou em acetato de celulose, permite a separação da Hb Fetal e sua quantificação densitométrica, fato que auxilia o diagnóstico dos diferentes genótipos das doenças falciformes. Por outro lado, a eletroforese alcalina em acetato de celulose é fundamental na identificação da Hb H e da metaemoglobina (que apresenta cor marrom) quando associadas à anemia falciforme. A dosagem de Hb Fetal é importante não apenas para caracterizar o genótipo da doença falciforme, mas principalmente para o entendimento da evolução clínica do paciente, onde o maior nível de Hb Fetal representa melhor prognóstico da doença. Por outro lado, a dosagem da metaemoglobina indica o grau de oxidação da Hb S, pois as concentrações elevadas estão relacionadas com processos fisiopatológicos mais deletérios ao eritrócito falciforme, bem como podem indicar a associação da Hb SS com deficiência de G-6-PD; nestes casos se faz necessário a dosagem enzimática desta enzima. A contagem de reticulócitos estabelece importantes parâmetros sobre o processo hemolítico decorrente da anemia falciforme, bem como a resposta da reposição eritrocitária – ou eritropoiese. A pesquisa intraeritrocitária de Hb H (figura 6.35) é um excelente método de confirmação da talassemia alfa. A pesquisa intraeritrocitária de corpos de Heinz representa importante informação sobre o desencadeamento de processos oxidativos da Hb S, bem como a suspeita da associação entre Hb SS e deficiência de G-6-PD.
Finalmente, é sempre importante enfatizar que para a correta conclusão do diagnóstico laboratorial da Hb SS ou da Hb SS/Talassemia alfa é necessário que se faça a análise de hemoglobinas no sangue dos pais. No caso da Hb SS, os genótipos mais comuns dos pais são: AS x AS; AS x SS; AS x SC; AS x S/Talassemia Beta. Quando a Hb SS está associada à talassemia alfa um dos pais deve apresentar a Hb H, por exemplo: AS x ASH; ASH x SC, etc.
