Autores:ppppPaulo Cesar Naoum Flávio Augusto Naoum |
||||||||||||||||||||||||||||||||||||||||||
Introdução Para descrever o fenômeno da falcização e as conseqüências advindas das células falcizadas é necessário recorrer ao eritrócito normal como parâmetro de comparação. O processo evolutivo que resultou na formação do eritrócito como célula especializada no transporte de gases, fez com que esta célula se tornasse discóide, bicôncava e flexível para realizar com eficiência as trocas gasosas e a permeabilidade de água e compostos iônicos. As milhões de moléculas de hemoglobinas contidas no interior de cada eritrócito normal se dispõe individualizadas e, portanto, solubilizadas no líquido intraeritrocitário. Todos esses fatores mantém a estrutura celular, e em especial a membrana citoplasmática, saudáveis durante cerca de 120 dias.Nos genótipos que causam a doença falciforme, e notadamente na anemia falciforme (Hb SS), as células falciformes tem suas origens determinadas na fase inicial da eritropoiese quando os proeritroblastos doentes tem em sua composição genética a lesão no gene da globina beta do cromossomo 11 (b6 Glu ® Val), sintetizando a Hb S. Portanto, durante toda a transformação e evolução das diferentes fases dos eritroblastos, as moléculas de Hb S são sintetizadas gradualmente e se ocupam do espaço intraeritrocitário. Conclui-se, assim, que as lesões dos eritrócitos falciformes tem início nas primeiras fases da sua formação, e esse fato pode ser constatado pela presença de alguns eritroblastos falcizados em sangue coletado por punção de medula óssea. Todo esse processo que ocorre de forma gradual e cumulativa, causa múltiplas alterações nas moléculas de Hb S, com disfunções celulares motivadas por alterações morfológicas e funcionais desses eritrócitos, reduzindo drasticamente seu tempo de vida média para 7 a 25 dias (figura 6.15). Embora todos os eventos que causam a falcização nos eritrócitos contendo moléculas de Hb S com concentrações acima de 50% ocorram concomitantemente, para fins didáticos eles serão expostos em três níveis: molecular, celular e circulatório. |
||||||||||||||||||||||||||||||||||||||||||
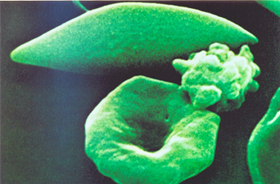
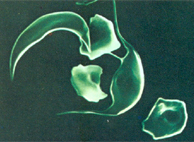
Figura 6.15 – Alterações morfológicas em eritrócitos de pessoa com anemia falciforme, por microscopia eletrônica. O eritrócito não falcizado apresenta várias lesões morfológicas de membrana devido à falcizações e desfalcizações ao longo do seu período de vida. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||

Figura 6.16 – Disposição gráfica da molécula de Hb S com seu tetrâmero a2 (verde) e b2 (azul). Os contatos a1 b1 e a2 b2 são verticais e estão relacionados à estabilidade. Entre os contatos b1 b2 está acomodada a molécula de 2,3 DPG que regula a oxigenação da molécula de hemoglobina. |
||||||||||||||||||||||||||||||||||||||||||
A base da fisiopatologia da falcização está na substituição do ácido glutâmico pela valina na posição 6 da globina beta. Embora esta mudança se configura bioquímica e geneticamente como pontual, numa região da molécula (superfície externa) que não a compromete estruturalmente, passa a ser suscetível quando a molécula de Hb S perde o oxigênio, tornando-se desoxi-Hb S. À medida que as moléculas de Hb S se tornam desoxigenadas dentro do eritrócito, ocorrem mudanças estruturais típicas de moléculas de hemoglobinas desoxigenadas, que no caso da Hb S tem conseqüências graves à sua individualidade molecular. Ao se tornarem desoxigenadas, as moléculas de Hb S expõe componentes carbonados da valina mutante (b 6 Val) que se interagem com componentes químicos da fenilalanina da posição 85 da globina beta (b 85 Fen), bem como com a leucina da posição 88 da mesma globina (b 88 Leu). Embora essas interações propiciam a ocorrência de outros contatos entre tetrâmeros de diferentes moléculas de Hb S, é justamente as interações b 85 Û b6 Û b 88 que dão o início à agregação entre moléculas de Hb S desoxigenadas. Quando 30 tetrâmeros estão agregados, é formado um núcleo crítico, que constitui a primeira fase de nucleação. Sobre este núcleo crítico, outros tetrâmeros podem aderir e formar então um polímero. Um polímero é uma estrutura longitudinal medindo 220nm, de diâmetro helicoidal, constituída por 14 fibras, 10 delas dispostas externamente e 4 internamente (figuras 6.17 a e 6.17 b). |
||||||||||||||||||||||||||||||||||||||||||

Figura 6.17 a – À esquerda a representação mostra que moléculas de Hb A ao perderem o oxigênio mantêm-se individualizadas. À direita a representação mostra que as moléculas de Hb S ao se tornarem desoxigenadas se agregam, dando início Pa formação de polímeros. |
||||||||||||||||||||||||||||||||||||||||||

Figura 6.17 b – À esquerda a representação mostra que milhares de moléculas de Hb S desoxigenadas se agregam e formam feixes de polímeros. À direita, a microscopia eletrônica plana de parte de um eritrócito falcizado mostra a disposição desses feixes de polímeros no sentido longitudinal da célula. |
||||||||||||||||||||||||||||||||||||||||||
Os polímeros estáveis fornecem uma área necessária para a iniciação de uma segunda fase de nucleação, na qual fibras adicionais podem ser agregadas, formando feixes paralelos, chamados tactóides. A dupla nucleação é responsável pelo crescimento exponencial dos polímeros e transformação do eritrócito discóide na forma afoiçada, conforme mostram as figuras 6.18, 6.19 e 6.20. Quando os polímeros adquirem uma configuração espacial definida, ocorre a mudança do estado líquido (ou solúvel) para o sólido (ou insolúvel) no interior do eritrócito, fato que altera a viscosidade do líquido intracelular e induz a formação de cristais de Hb S. |
||||||||||||||||||||||||||||||||||||||||||
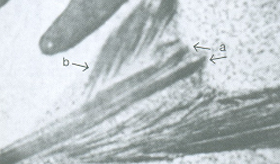
Figura 6.18 – Microscopia eletrônica de um eritrócito com Hb S no início da formação da polimerização de moléculas de desoxi-Hb S. As setas a indicam dois grandes polímeros longitudinais estáveis, e a partir deses ocorre a nucleação com fibras adicionais de feixes paralelos, os tactóides (beta b). |
||||||||||||||||||||||||||||||||||||||||||

Figura 6.19 – Na seqüência da figura anterior, milhares de polímeros são formados, conforme mostra esta imagem de microscopia eletrônica de uma célula falcizada. Há duas áreas bem distintas formadas pelos polímeros longitudinais estáveis (a) e a nucleação com fibras adicionais (b). |
||||||||||||||||||||||||||||||||||||||||||
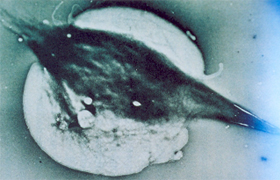
Figura 6.20 – Fase final da polimerização com a transformação do eritrócito discóide em célula falciforme, observada em microscopia eletrônica. O processo imediato da falcização deixa um espaço vazio em forma de disco que era ocupado pelo eritrócito com oxi-Hb S, para se condensar na estrutura afoiçada induzida pela polimerização das moléculas de deoxi-Hb S. |
||||||||||||||||||||||||||||||||||||||||||
A polimerização de moléculas de Hb S solubilizadas "in vitro" é dependente do oxigênio, do pH, da concentração do 2,3 DPG, da temperatura, da concentração da Hb S e das interações com a Hb Fetal e outras hemoglobinas variantes. O oxigênio é o elemento mais importante, pois somente a desoxi-Hb S se polimeriza, e qualquer fator que estabilize o estado desóxi reduzirá a afinidade pelo oxigênio, perpetuando o estado desóxi e a polimerização. Entre os fatores que mantém o estado desoxigenado se destacam a queda do pH pelo efeito Bohr e o aumento da concentração do 2,3 difosfoglicerato. Da mesma forma, o aumento da temperatura acima de 37ºC induz a polimerização da Hb S, bem como provoca a sua instabilidade molecular. A concentração da Hb S corpuscular média (CHCM) é fundamental no desencadeamento da polimerização, pois quanto maior a presença de Hb S intraeritrocitária menor será o tempo de formação de polímeros e, consequentemente, a polimerização se torna mais rápida. Admite-se que a polimerização que ocorre "in vivo" parece ter igual comportamento daquela que se verifica "in vitro", entretanto há grandes diferenças entre uma e outra. Ou seja, no processo "in vivo", o espectro de células com diferentes densidades e concentração de Hb S é variável entre os diversos genótipos que causam as doenças falciformes, além do que há diferenças dentro de um mesmo genótipo – quer seja pela presença de Hb Fetal ou pela associação de talassemia alfa, entre outros. Essas diferenças promovem variações entre o tempo do processo polimerizante que varia de milisegundos a muitos segundos. Soma-se a essas situações "in vivo" aos micro-ambientes em que os eritrócitos com Hb S estão circulando, em determinado momento. E esse fato pode ser atestado nos processos vaso-oclusivos que ocorrem com mais intensidade em certas regiões do corpo humano. Mutações adicionais no gene da globina beta S e a herança de outras hemoglobinas variantes associadas à Hb S podem afetar a estabilidade dos polímeros de Hb S, aumentando-a, ou diminuindo-a, ou permanecendo a mesma tendência do efeito polimerizante. O aumento da intensidade da polimerização depende da concentração da Hb S desoxigenada, assim é evidente que na Hb SS com concentrações de Hb S próximas de 95 a 98% o efeito polimerizante é altíssimo. Nas associações entre Hb S e Hb D Punjab (Hb SD), e entre Hb S e Hb O Arábia (Hb SO), a polimerização é quase tão intensa quanto na Hb SS, e os fenótipos das doenças falciformes SD e SO são muito parecidos com o da anemia falciforme. Entretanto quando a associação da Hb S se faz com a Hb E (Hb SE), ou com Hb Korle-Bu (Hb S/Korle-Bu), o grau de polimerização é muito pequeno, assemelhando-se com o traço falcêmico. Na associação entre Hb S e Hb C a polimerização é moderada, com características laboratoriais como, por exemplo, a formação de cristais intra-eritrocitários. A interação da Hb S com a Hb Fetal é determinante na diminuição do efeito polimerizante da Hb S. A Hb Fetal não é incorporada no polímero da Hb S e por essa razão a polimerização da Hb S é inibida. Esse processo inibitório da polimerização da Hb S promovido pela presença de Hb Fetal ocorre na persistência hereditária de Hb Fetal (PHHF), situação em que a concentração da Hb Fetal oscila entre 15 e 30%. Nesses casos o portador do genótipo SF – onde a Hb Fetal se deve à PHHF – dificilmente apresenta o fenótipo da doença falciforme, assemelhando-se mais com o traço falciforme. Essa relação entre polimerização e fenótipo clínico é o que sustenta a hipótese da importância da concentração de Hb Fetal na fisiopatologia da doença falciforme. |
||||||||||||||||||||||||||||||||||||||||||
|
A polimerização
da Hb S começa a deformar o eritrócito imediatamente,
fazendo com que a célula adquira a forma alongada –
inicialmente com seu diâmetro central visivelmente achatado
– tipo "lâmina de foice", culminando com o
aparecimento de longos filamentos nas extremidades. Na realidade,
as alterações observadas nas células falciformes
são muito variadas (figura 6.21). A redução
da deformabilidade dos eritrócitos falcêmicos é
uma das principais causas das conseqüências fisiopatológicas
das doenças falciformes. Contribui para este fato, inicialmente,
a formação dos polímeros de Hb S, seguida das
alterações da membrana celular e do seu citoesqueleto,
além dos produtos de degradação oxidativa da
hemoglobina, caracterizadas pelos corpos de Heinz. Entre as principais
conseqüências das lesões à membrana e ao
citoesqueleto destaca-se a desidratação celular, seguida
da ação macrofágica contra os eritrócitos
que sofreram oxidação da proteína Banda 3 das
suas membranas, fatos que alteram a composição estrutural
da membrana do eritrócito falciforme. |
||||||||||||||||||||||||||||||||||||||||||
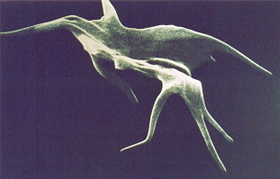 Figura 6.21 – Microscopia eletrônica de células falciformes com diferentes formas e filamentos nas extremidades. |
||||||||||||||||||||||||||||||||||||||||||
A desidratação celular ocorre devido à disfunção da permeabilidade da célula falciforme motivada pela falência parcial da bomba de sódio/potássio/ATPase, resultando na perda de potássio e ganho de sódio que, se balanceado, não altera a hidratação celular, mas, se houver desequilíbrio, haverá perda excessiva de potássio e de água, com aumento da concentração intracelular de Hb S e conseqüente polimerização. A perda de potássio é a principal causa da desidratação dos eritrócitos falciformes. Os fatores que mais contribuem para a perda de potássio são o co-transporte de K+Cl–, e a formação dos canais de Gardos (Ca++ atividade dependente de K+). O co-transporte de K+Cl– é ativado pelo edema celular e por acidose. Esta pode ocorrer, "in vivo", particularmente em condições de circulação estagnante. A concentração intraeritrocitária de Hb S está diretamente relacionada com o conteúdo de Ca++, e este é compartimentalizado em vesículas intracelulares, mantendo a concentração de Ca++, no citosol, normal. Quando há distorção da membrana, como no processo de falcização, ocorre aumento da permeabilidade da membrana celular ao cálcio, elevando a sua concentração no citoplasma (figura 6.22). |
||||||||||||||||||||||||||||||||||||||||||
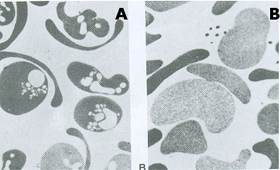
Figura 6.22 – Microscopia eletrônica de eritrócitos normais com Hb AA (A) e falciformes (B) tratados in vitro para avaliar sua permeabilidade do cálcio e seu fluxo. Em eritrócitos normais o efluxo de cálcio é muito maior, caracterizado pelas vesículas intra-eritrocitárias (A), enquanto nos eritrócitos falcêmicos a permeabilidade é maior para o influxo, tornando-os densos. |
||||||||||||||||||||||||||||||||||||||||||
Como os eritrócitos tem pequena capacidade de tampão para o cálcio, a mínima entrada deste elemento induz o aumento significativo da sua concentração citoplasmática. Assim, para manter o gradiente de equilíbrio no citosol da célula, os canais de Gardos são ativados, e desta forma permite a entrada de potássio na célula. Quando o potássio entra na célula, a membrana celular torna-se eletricamente mais negativa, fazendo com que íons cloretos (Cl–) saiam da célula, juntamente com o potássio. Todas essas alterações funcionais que ocorrem na célula falciforme, desencadeada inicialmente pela desoxigenação da Hb S, seguida pela formação de polímeros, causam alterações na permeabilidade da membrana, com lesões do citoesqueleto celular (figura 6.23) e promovendo a rigidez da membrana eritrocitária. |
||||||||||||||||||||||||||||||||||||||||||

Figura 6.23 – Microscopia eletrônica do citoesqueleto de um eritrócito normal com Hb AA (esquerda) e um eritrócito falciforme com Hb SS (direita). Observar a desorganização estrutural do citoesqueleto da célula falciforme em relação ao eritrócito normal. |
||||||||||||||||||||||||||||||||||||||||||
Embora esses fenômenos possam ser reversíveis com a reoxigenação, quando repetidos, as alterações funcionais exacerbam-se e a célula se torna irreversivelmente falcizada. O tempo de falcização, até a distorção celular acentuada, é variável entre dois a quatro minutos. O tempo de contato dos eritrócitos com a circulação venosa é de 10 a 15 segundos, portanto em áreas de estagnação do sangue a vulnerabilidade à falcização é maior. No entanto, a circulação venosa tem maior número de eritrócitos falciformes irreversíveis do que a arterial, porque o pré-requisito essencial seria o aumento da CHCM, causado pela desidratação celular e pela falência da atividade funcional da célula na circulação, estabilizando-se o esqueleto celular na forma anormal. A vasoconstrição da circulação capilar induz a estase e ao aumento do tempo de contato do eritrócito com áreas de baixo teor de oxigênio. Também a desidratação do organismo favorece a desidratação celular e o aumento da concentração de Hb S. Os homozigotos falcêmicos (Hb SS) – cujo fenótipo é o da anemia falciforme – possuem de 4 a 44% de eritrócitos falciformes irreversíveis na circulação periférica. Seu número é relativamente constante no mesmo indivíduo, apesar de variar de um caso para outro, e esse processo está relacionado com a vida média celular determinada pelo grau de hemólise. Conforme mostra a figura 6.24, três eventos principais participam da formação da célula falciforme: a desoxigenação da Hb S – como indutor – durante a qual são liberados radicais livres oxidantes, a formação de polímeros – cuja abordagem foi feita no início deste capítulo, e a degradação oxidativa da Hb S – iniciada pela metaemoglobinização da Hb S e formação de corpos de Heinz. Todos esses eventos causam lesão e rigidez da membrana do eritrócito. Decorrente dessas alterações da célula falciforme é importante destacar especificamente a disfunção estrutural da membrana e a degradação oxidativa da Hb S. |
||||||||||||||||||||||||||||||||||||||||||
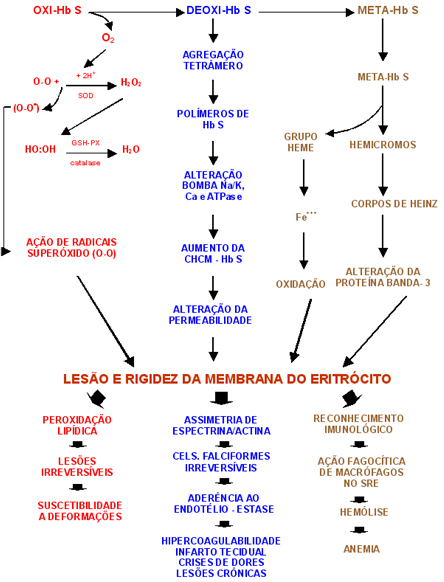 Figura 6.24 – Conseqüências celular e fisiopatológica da desoxigenação da Hb S. |
||||||||||||||||||||||||||||||||||||||||||
A membrana celular é
uma bicamada de fosfolipídeos medindo cerca de 10nm de espessura.
Os principais componentes são lipídios e proteínas.
Os três principais lipídios são: os fosfolipídios,
colesterol e glicolipídios. Os fosfolipídios são
anfipáticos, isto é, possuem uma cabeça polar
hidrofílica que interage fortemente com a água e uma
cauda hidrofóbica que interage, por sua vez, com a outra
porção hidrofóbica da bicamada. Com esta propriedade
anfipática, os fosfolipídios em meio aquoso, formam
espontaneamente uma bicamada planar, pois esta é a configuração
de menor gasto de energia. A membrana celular dos eritrócitos e também de outras células, apresenta uma distribuição assimétrica dos fosfolipídios. Na camada externa da membrana celular está 75% a 80% do total de fosfolipídios; estes contém colina e são: fosfatidilcolina e esfingomielina; o restante, 20% a 25% dos fosfolipídios, está na superfície interna da membrana celular, e são os aminofosfolipídios: fosfatidilserina e fosfatidiletanolamina (figura 6.25). |
||||||||||||||||||||||||||||||||||||||||||
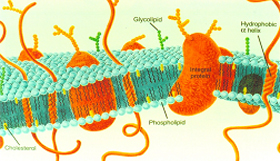
Figura 6.25 – Disposição química de membrana normal de eritrócitos com destaques para fosfolipídeos, proteína integral, colesterol, glicolipídeo e proteínas hidrofóbicas. |
||||||||||||||||||||||||||||||||||||||||||
Uma molécula lipídica polar, na superfície de uma membrana, é livre para se mover no seu próprio lado da bicamada, mas é impedida de deslocar-se para a superfície do outro lado. A resistência da bicamada à transferência de lipídios de uma face da membrana para outra mantém e preserva a assimetria da bicamada. Esta resistência é causada pela grande quantidade de energia necessária para empurrar as regiões polares dos fosfolipídios ao longo da base hidrocarbonada da membrana. Outros mecanismos envolvidos na estabilidade dos fosfolipídios são: a) a interação da fosfatidilserina com proteínas do citoesqueleto celular, como a espectrina e Banda 4.1 e b) a ativa manutenção da orientação por uma proteína de translocação de fosfolipídios ATP dependente, a aminofosfolipídio transferase (figura 6.26). |
||||||||||||||||||||||||||||||||||||||||||
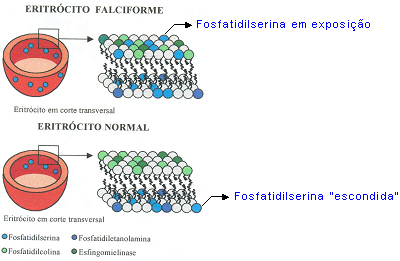
Figura 6.26 - O eritrócito falciforme apresenta em sua composição de membrana a exposição de fosfatidil serina na superfície externa da bicamada, enquanto que no eritrócito normal a fosfatidil serina está na superfície interna da bicamada. A exposição da fosfatidil na superfície externa ocorre durante a desoxigenação e sua composição química contribui com os processos de vaso-oclusão pelo aumento da aderência das células falciformes ao endotélio vascular. |
||||||||||||||||||||||||||||||||||||||||||
A relação entre os lipídios da camada interna e externa é o maior determinante da forma discóide dos eritrócitos. O transporte ativo de íons e água através da membrana é necessário para manter o equilíbrio de lipídios nas monocamadas, bem como para recuperar sua forma normal após a deformação. Alterações na forma dos eritrócitos, tal como se verifica na falcização, são acompanhadas de uma troca de relação de área da camada externa com a camada interna. A reversibilidade da deformação que um eritrócito sofre, após passar pela microcirculação, depende de um rápido fluxo de lipídios de uma lado para outro, implicando que a deformabilidade da célula é influenciada pela taxa de difusão transversal de lipídios. A assimetria dos fosfolipídios é essencialmente conservada por toda a vida da célula, e se mantém em equilíbrio dinâmico na estrutura da membrana. Todos os fosfolipídios se difundem passivamente através da bicamada, em uma taxa lenta. Os aminofosfolipídios são transportados ativamente pela enzima aminofosfolipídio-transferase, sendo esta responsável pelo rápido transporte da fosfatidilserina para a monocamada interna. Em situações em que a atividade da aminofosfolipídio-transferase está prejudicada, como ocorre em sangue estocado, não se observa a perda da assimetria, pois o movimento da fosfatidilserina para a camada externa é extremamente lento. Entretanto, a perda da assimetria é observada quando além da alteração na atividade da enzima, existe também lesão da membrana que pode ser provocada "in vitro" por cálcio e ionofore, e "in vivo" pela formação de polímeros de Hb S. Diante dessas situações, e em particular do eritrócito falciforme, ocorre a exposição da fosfatidilserina na superfície externa da bicamada lipo-proteica da membrana. A exposição da fosfatidilserina contribui com os processos vaso-oclusivos (figura 6.27), aumentando a aderência das células falciformes ao endotélio vascular, levando ao estado de hipercoagulabilidade, infarto tecidual, crises de dores e lesões crônicas dos órgãos prejudicados. |
||||||||||||||||||||||||||||||||||||||||||
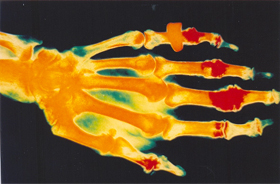
Figura 6.27 – Radiografia termográfica da mão de um paciente com anemia falciforme, mostrando a oclusão vascular nos dedos e a estagnação de sangue. |
||||||||||||||||||||||||||||||||||||||||||
As lesões celulares
causadas por radicais livres derivados de espécies ativadas
de oxigênio (O2•, H2O,
HO) estão associadas com a diminuição da sobrevida
do eritrócito em vários tipos de anemias hemolíticas,
quer sejam de origem adquirida, hereditária, ou induzida
por drogas oxidantes. A produção de espécies
ativadas de oxigênio e a suscetibilidade da oxihemoglobina
à autoxidação relacionam-se com a capacidade
de um elétron do ferro se despolarizar no momento em que
o oxigênio é liberado do grupo heme. Essa suscetibilidade
depende da capacidade estrutural do grupo heme e dos aminoácidos
que o protegem para manter o ferro no estado ferroso e, assim, torna-lo
hábil para se ligar reversivelmente ao oxigênio. Qualquer
modificação no pacote do grupo heme – mesmo
que seja de pequenas dimensões – pode permitir o acesso
de ânions ou moléculas de água para o seu interior
e deslocar um elétron do ferro, fato que resultará
na formação de radicais superóxidos (O2•
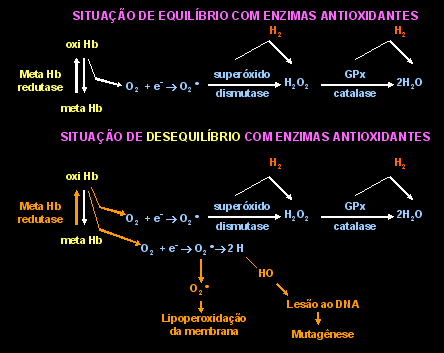
ou O - O• ) e de metahemoglobina (figura 6.24). Em eritrócito normal é comum a ocorrência de autoxidação espontânea da hemoglobina que induz a geração de 107 radicais superóxidos dentro da célula. Esses radicais são facilmente controlados pelas defesas antioxidantes dos eritrócitos. Qualquer situação patológica que aumenta a geração de radicais superóxidos, quer seja pela elevação de estresse oxidativo ou pelo desequilíbrio das defesas antioxidantes, acentuará a produção de espécies ativadas de oxigênio. Entre as principais situações de patologias eritrocitárias hereditárias destacam-se a anemia falciforme e as implicações desses radicais livres na lipoperoxidação da membrana do eritrócito falciforme, a talassemia maior no qual o excesso de ferro extracelular e a globina despareada formam complexos residuais que também lesam a membrana, e a deficiência de glicose-6-fosfato desidrogenase (G6PD) cuja oxidação da hemoglobina e conseqüente lesão da membrana se verifica pela deficiência de mecanismos de detoxificação do peróxido de hidrogênio (H2O2). A geração de radicais livres nos eritrócitos com Hb S ocorre quando a oxi-Hb se muda para a forma desoxi-Hb (figura 6.24). A liberação do oxigênio o torna suscetível ao ataque de elétrons que estão no interior da célula. Para evitar que o oxigênio fique eletrizado e se transforme em íon superóxido, a enzima antioxidante superóxido dismutase (SOD) atua no sentido de transformar o radical superóxido para peróxido de hidrogênio. O peróxido de hidrogênio, que também é um radical livre, sofre a ação antioxidante de outras duas enzimas eritrocitárias: a glutationa peroxidase (GSH-Px) e a catalase, transformando-o em água. O esquema abaixo mostra a geração e os efeitos deletérios dos radicais livres durante a desoxigenção da hemoglobina. Esse processo é mais intenso quando a Hb S intraeritrocitária tem concentração superior a 50%. |
||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||
A deficiência hereditária de enzimas antioxidantes tem sido relatada em diversas populações, e quando associada especialmente à anemia falciforme contribui ainda mais na oxidação da Hb S, elevando a formação de produtos de degradação desta hemoglobina, fatos que causam a redução da vida média do eritrócito falcêmico, aumento do grau de hemólise e intensificação da anemia. Como foi apresentado há pouco, as proteínas da membrana eritrocitária podem ser alvos dos ataques de radicais livres quando estes estão dentro ou fora das células em altas concentrações, desnaturando-as. O acúmulo de proteínas desnaturadas interfere nas funções celulares. Vários aminoácidos de importância para a função protéica são suscetíveis às lesões causadas pelos radicais livres: metionina, triptofano, histidina, cisteína e tirosina. As proteínas lesadas por radicais livres podem dar origem a outros radicais, notadamente aqueles classificados como espécies ativadas de oxigênio, e em especial o radical hidroxila (HO) gerado pelo acúmulo de H2O2 na presença de ferro proveniente da degradação da Hb S. Entre os radicais intermediários destacam-se os hidroperóxidos lipídicos que ao reagirem com o ferro geram os radicais alcoxil (RO•·) e peroxil (ROO•·). A geração desses radicais amplificam as lesões celulares devido aos contínuos processos de novos ciclos de peroxidação lipídica, resultando na liberação de alcanos (malonaldeídos) e alquenos (4-hidroxinonenal), causando lesões irreversíveis nos eritrócitos falcizados e tendência a deformações morfológicas com implicações fisiopatológicas. Como se pode observar, todos os efeitos deletérios dos eritrócitos com Hb S ocorrem a partir da transformação do estado oxi-Hb S para desoxi-Hb S, desencadeando três processos fisiopatológicos caracterizados pela formação de polímeros de Hb S, degradação da meta-Hb S, e liberação de espécies ativadas de oxigênio ou radicais livres. Os produtos e subprodutos provenientes dessas cadeias de reações atacam a membrana do eritrócito com Hb S, provocando lesões em sua estrutura que gradualmente reduzem o grau de deformabilidade tornando-a rígida. As conseqüências advindas da desestruturação funcional e morfológica do eritrócito falciforme também estão resumidas na figura 6.24. Os aspectos práticos da presente exposição podem ser fundamentados em resultados de pesquisas obtidos da literatura, bem como em nosso laboratório. Das e Essman demonstraram que em pacientes com anemia falciforme os mecanismos antioxidantes dos eritrócitos estavam defeituosos devido à diminuição das atividades de glutatião peroxidase (GSH-Px) e catalase, e aumento dos níveis de superóxido dismutase (SOD). Similares alterações também foram descritas em outras doenças que envolvem mecanismos oxidativos, como são os casos da talassemia beta maior e da deficiência de G-6PD. Rice-Evans e colaboradores sugeriram que a diminuição da atividade de GSH-Px poderia ser responsável pela acelerada peroxidação lipídica da membrana eritrocitária e, consequentemente, induzir a anemia hemolítica. Essa suposição foi confirmada em trabalhos posteriores ao demonstrarem que os eritrócitos sob estresse oxidativo, seja de origem induzida ou constitucional, apresentam maior grau de hemólise e a sua intensidade está relacionada com o local em que a célula sofreu a agressão. Trabalhos realizados em nosso laboratório mostraram que há direta relação entre o aumento da concentração de Hb S com a elevação de metahemoglobina, conforme resultados obtidos em amostras de sangue com Hb AS, SS e S/Talassemia beta (tabelas 6.5 e 6.6). Observa-se pela análise da tabela 20 que o nível médio da concentração de metaemoglobina está aumentado nos genótipos que tem a presença de Hb S. Por outro lado, a análise da tabela 21 torna evidente que a oxidação espontânea da Hb SS e Hb S/ b talassemia ocorre em mais de 50% dos portadores desses genes, uma vez que a concentração de metaemoglobina está elevada acima de 2%. Entretanto, é no genótipo SS que a oxidação da Hb S é maior, pois, 35,29% dos pacientes analisados apresentaram concentrações de metaemoglobina superior a 5%. A avaliação da precipitação de corpos de Heinz em eritrócitos com Hb S foi efetuada comparativamente com os valores de concentrações desta hemoglobina (<50%, 51 a 80%, e acima de 80%) e confrontada com a Hb AA (tabela 6.7). Os resultados caracterizaram com muita clareza que a presença de corpos de Heinz é mais intensa quanto maior a concentração de Hb S. Por essas análises (concentração de metaemoglobina e corpos de Heinz) é possível avaliar o potencial oxidativo de determinada amostra de sangue com Hb S. Quanto maior o potencial oxidativo, maior serão também as conseqüências fisiopatológicas para as células falciformes. Assim, os resultados laboratoriais das doenças falciformes contendo as análises do potencial oxidativo se mostram como parâmetros eficientes para avaliarem os procedimentos médicos adequados aos pacientes falcêmicos. |
||||||||||||||||||||||||||||||||||||||||||
Tabela 6.5 - Análise descritiva (número de amostras analisadas, média, mediana, valores mínimo e máximo, desvio padrão e erro padrão) da concentração percentual de metaemoglobina (MetaHb) para os genótipos AA, AS, SS e S/ b tal. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Tabela 6.6 - Distribuição percentual em três faixas de concentração de metaemoglobina (MetaHb) para os genótipos AA, AS, SS e S/ b tal. |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Tabela 6.7 - Análise descritiva (número de amostras analisadas, média, mediana, valores mínimo e máximo, desvio padrão e erro padrão) da quantidade de eritrócitos com corpos de Heinz por 1000 eritrócitos analisados e relacionada com valores percentuais da concentração de Hb S e do controle (Hb AA). |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||

Figura 6.28 – Microscopia eletrônica de corte transversal de uma artéria. A parede espessa e elástica dessa artéria suporta a alta pressão do fluxo sanguíneo. |
||||||||||||||||||||||||||||||||||||||||||
O segundo compartimento é formado por arteríolas cujas contrações podem afetar a circulação e distribuição do sangue, e definem a pressão capilar pós-arteriolar. Este compartimento tem paredes vasculares capazes de suportar mudanças rápidas da pressão arterial (5 a 15 Pa) em frações de segundos, e de se adaptar ao volume de viscosidade sangüínea (figura 6.29). |
||||||||||||||||||||||||||||||||||||||||||
 Figura 6.29 – Microscopia eletrônica do endotélio (parede interna) de uma arteríola do glomérulo renal que suporta mudanças rápidas da pressão arterial e se adapta ao volume da viscosidade sanguínea. |
||||||||||||||||||||||||||||||||||||||||||
O terceiro compartimento é formado por extensa rede de veias capilares e pós-capilares, com elevada capacidade superficial para suportar o volume de sangue do sistema circulatório (figura 6.30). |
||||||||||||||||||||||||||||||||||||||||||
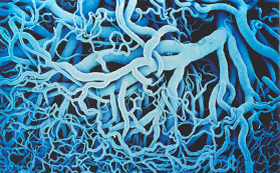
Figura 6.30 – Microscopia óptica da rede capilar de uma parte do pulmão. O grande volume de superfície vascular permite realizar excepcional volume de troca gasosa de suas paredes. |
||||||||||||||||||||||||||||||||||||||||||
O diâmetro capilar pode ser menor que o diâmetro de um eritrócito, e requer, por isso, grande capacidade de deformação desta célula (figura 6.31). |
||||||||||||||||||||||||||||||||||||||||||
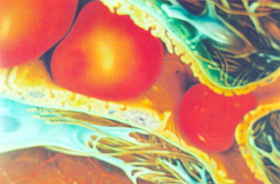
Figura 6.31 – Figura ilustrativa em dimensões obtidas pela microscopia eletrônica de varredura de um capilar extremamente delgado, onde o eritrócito se deforma ara poder fluir na circulação. |
||||||||||||||||||||||||||||||||||||||||||
O quarto compartimento é composto por vasos venosos de tamanhos variáveis entre médio e grande, capazes de suportarem em determinados momentos até 80% do volume total de sangue e exercem baixas pressões sobre suas paredes (menos de 1 Pa). A circulação do sangue tem características especiais conforme variam os diferentes tamanhos de vasos sangüíneos. Em vasos com diâmetros superiores a 300mm o fluxo de sangue ocorre com viscosidade moderadamente baixa, e os eritrócitos se dispõem em grupos agregados (figura 6.28). Por outro lado, o fluxo em capilares sangüíneos entre 6 e 12mm faz com que as células se disponham em "fila indiana" (figura 6.32), com intensa interação entre os eritrócitos com as células endoteliais que compõem as paredes dos vasos capilares. |
||||||||||||||||||||||||||||||||||||||||||
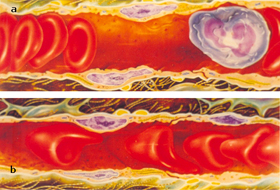
Figura 6.32 – Figura ilustrativa em dimensões obtidas ela microscopia eletrônica de varredura: (a) em circulação de baixa velocidade os eritrócitos têm a forma discóide; (b) em circulação de alta velocidade os eritrócitos se deformam adquirindo formas que lembram “pára-queda”, “projétil”, etc. |
||||||||||||||||||||||||||||||||||||||||||
| Em circulação com baixa velocidade nesses vasos, os eritrócitos se apresentam com a forma discóide (figura 6.32 a), fato que não se observa quando o fluxo sanguíneo se apresenta em alta velocidade onde os eritrócitos se deformam, adquirindo a forma de "pára-quedas" (figura 6.32 b). A circulação sangüínea de eritrócitos falciformes se destaca inicialmente devido às desigualdades morfológicas dessas células. Essa desfiguração eritrocitária influi intensamente no fluxo sangüíneo da microcirculação pois, a irregularidade da superfície de contato da célula falciforme, permite reações químicas interativas entre os eritrócitos falcêmicos e as células endoteliais, fazendo-as aderirem ao endotélio vascular. As conseqüências da aderência é caracterizada pela vaso-oclusão, com redução do fluxo sangüíneo nos capilares venosos, causando a estase venosa e hipóxia. Na realidade essas lesões que acontecem na microcirculação de vários órgãos e tecidos, determinam um fenômeno cíclico que, iniciado pela falcização, esse mesmo processo é continuadamente estimulado pelos seus próprios efeitos deletérios conforme mostra esquema abaixo. |
||||||||||||||||||||||||||||||||||||||||||
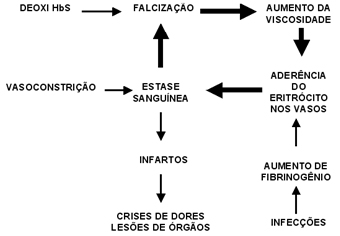
Esquema ilustrativo da seqüência das lesões que ocorrem na microcirculação sangüínea. |
||||||||||||||||||||||||||||||||||||||||||
A abrangência orgânica da vaso-oclusão depende de outras situações que naturalmente se somam, quais sejam, suscetibilidades a infecções bacterianas e virais que induzem o aumento de fibrinogênio e estimulam a aderência da célula falciforme ao endotélio, eventos que promovem vasoconstrição, e a própria hipóxia tecidual. É justamente a hipóxia tecidual que promove a continuidade da desoxigenação de moléculas de Hb S, agravando ainda mais uma condição circulatória já desfavorável e lesando os tecidos perfundidos por esses capilares. Ocasionalmente pode ocorrer oclusão total dos capilares com trombose, formação de fibrina com a participação das plaquetas ativadas pela assimetria anormal da estrutura da membrana do eritrócito falcêmico, e também ativação dos fatores de coagulação – propiciando a hipercoagulabilidade. Os tecidos malperfundidos podem sofrer infartos com necrose e formação de fibrose, principalmente no baço, na medula óssea e placenta. Todos esses processos fisiopatológicos são causas importantes de lesões teciduais agudas – caracterizadas pelas crises de dores, e crônicas – situações determinantes do alto grau de morbidade e mortalidade notadamente na anemia falciforme. Destaque importante no das doenças falciformes, e em especial da anemia falciforme, se deve ao conhecimento sobre a fisiopatologia da vaso-oclusão. |
||||||||||||||||||||||||||||||||||||||||||
A vaso-oclusão nas doenças
falciformes pode ser explicada em quatro fases: iniciação,
extensão, lesão e reparo. Iniciação: é considerada a primeira fase da obstrução vascular, e se deve ao envolvimento da célula falcizada com todas as suas alterações morfo-funcionais relacionadas com a peculiaridade anatômica dos vasos em que estejam circulando naquele determinado momento. Dessa forma, as características vasculares associadas às deformações das células falcizadas, induzem mudanças profundas que alteram a fisiologia vasomotora bem como fatores humorais na região anatomicamente afetada, fatos que influenciam a tendência das células falciformes aderirem ao endotélio e provocar a oclusão vascular. Todos esses eventos acima descritos, que ocorrem simultaneamente ou sucessivamente, atuam como moduladores determinantes na causa da obstrução vascular. É importante destacar que o processo vaso-adesivo, determinado pela agregação das células falciformes entre si, e de suas adesões ao endotélio vascular, incluem, também, a participação de plaquetas, leucócitos e fatores hemostáticos. Apesar de não conhecer com certeza a forma e a intensidade desta participação, se sabe que a exposição da fosfatidilserina na superfície dos eritrócitos falciformes pode ser um fator iniciador ou de manutenção dos episódios de vaso-oclusão na microcirculação. Da mesma forma, os neutrófilos ativados pelas alterações que ocorrem na região em que se processa a fase de iniciação também se aderem ao endotélio e, além disso, se torna estruturalmente mais rígido e se unem também às células falciformes. Os monócitos também tem importante participação durante a oclusão vascular devido aos processos interativos com as células endoteliais da região, com intensa síntese de TNF-a (fator de necrose tumoral-a), IL-1 (interleucina-1) e TGF-b (fator de crescimento de células T-b). Esse fenômeno interativo induz as células endoteliais a liberarem IL-1 e IL-6, além de GM-CSF (fator estimulador de granulócitos e macrófagos). Essas citocinas (IL, GM-CSF, TNF e TGF) tem grande influência na síntese de anticorpos e de outros fatores humorais responsáveis pela febre, choque, etc. Os efeitos clínicos decorrentes dos processos vaso-oclusivos serão apresentados no item específico deste capítulo. A iniciação de um episódio de oclusão vascular também pode ocorrer como consequências de alterações que ocorrem no tono vascular, possivelmente resultante de mudanças nas reações que controlam os mecanismos metabólicos e neuro-hormonais, potencialmente induzidos por infecção ou inflamação, que anulariam os ajustes hemodinâmicos compensatórios necessários para o fluxo das células falciformes. Extensão: a modificação do fluxo sangüíneo no vaso obstruído pelas células falciformes agregadas e aderentes ao endotélio vascular promove a extensão dos eventos vaso-oclusivos, quer seja próximo ou distante do foco de iniciação. Lesão: as doenças falciformes, e em especial a anemia falciforme, são caracterizadas por episódios periódicos de eventos vaso-oclusivos, que ao longo do tempo causam lesões em vários órgãos do abdome, pequenos ossos da mão e pé, grandes ossos, coluna, articulações, vasos cerebrais, pulmões e baço, principalmente. Reparo: embora os múltiplos processos de lesões vaso-oclusivas têm efeitos progressivos em vários órgãos e, antes que muitos deles entram em falência fisiológica, ocorrem mecanismos de reparos no nível celular. Esses mecanismos estão relacionados principalmente com a qualidade da saúde de cada paciente, admitindo-se, portanto, que esses reparos podem ser eficientes ou insuficientes. A oclusão vascular promovida pelas células falciformes é uma situação que ocorre quando a concentração intracelular da Hb S é superior a 40%. Assim, é um processo quase comum entre os genótipos que caracterizam a doença falciforme: SS, SD, S/talassemia beta, SS/talassemia alfa, SC, e SS/PHHF. No traço falciforme, a oclusão e adesividade das células falciformes, é uma situação extremamente difícil de ocorrer. Entretanto, pelo fato da medula renal ser sempre acidótica, hipóxica e com hiperosmolaridade, essas condições favorecem a polimerização da Hb S, mesmo para os genótipos com concentrações abaixo de 40% como é o caso da Hb AS, principalmente. Muitas vezes, essas situações localizadas induzem os eritrócitos com Hb AS a se deformarem e causam, principalmente, a hipostenúria e, ocasionalmente, a hematúria. |
||||||||||||||||||||||||||||||||||||||||||
Pesquisas realizadas por Hebell
e colaboradores ao longo de quase dez anos, demonstraram que as
células falciformes tem aderência anormal ao endotélio.
Contribuem, para este fato, a exposição de fosfatidilserina
na membrana do eritrócito, a densidade celular, e a presença
de compostos químicos mediadores da aderência ao endotélio
vascular. A exposição da fosfatidilserina já
foi apresentada neste capítulo. Com relação
à densidade celular, se sabe que como conseqüência
da desidratação celular, existe uma heterogeneidade
das células falciformes que podem ser demonstradas por meio
da centrifugação em gradientes de densidades. A densidade
das células falciformes é determinada pela concentração
de hemoglobina corpuscular média (CHCM), onde a maior concentração
de Hb S determina maior gradiente de densidade. As células
com densidade alta são referidas como células densas.
Podem ser definidas quatro categorias de células baseadas
no gradiente de densidade por centrifugação. As quatro
categorias de células falciformes, ou SS, são as seguintes:
1) Células SS 1 – reticulócitos |
||||||||||||||||||||||||||||||||||||||||||
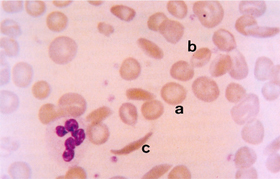
Figura 6.34 - Microscopia óptica de esfregaço obtido de amostra de sangue com diagnóstico de anemia falciforme (a) células com Hb S na forma discóide – SS 2; (b) --- momento inicial da polimerização caracterizada pela condensação de Hb S numa parte da célula discóide densa – SS 3; (c) célula falciforme irreversível – SS 4. |
||||||||||||||||||||||||||||||||||||||||||
Cada uma dessas categorias de células tem características morfológicas e propriedades distintas, contribuindo de forma diferente com a vaso-oclusão e hemólise. As células falciformes aderem exclusivamente ao endotélio venular e a aderência é inversamente relacionada ao diâmetro da vênula. Estudos com cultura de células endoteliais mostraram que, quanto maior a densidade do eritrócito falciforme, maior será o seu potencial de aderência. Porém, os estudos com as células durante o fluxo sangüíneo, mostraram que a aderência ao endotélio está inversamente relacionada ao CHCM. Assim, o processo de vaso-oclusão é iniciado com a aderência de células de baixa densidade ao sistema venular pós-capilar, diminuindo o fluxo sangüíneo local, e facilitando a aderência de células falciformes irreversíveis. Os mecanismos biológicos que promovem a iteração das células falciformes com as células endoteliais diferem entre os tipos de endotélio. As células falciformes se aderem às células endoteliais por meio da superfície de antígenos CD 36 e CD 44, integrinas e outros componentes específicos da membrana do eritrócito falciforme. Proteínas plasmáticas como o fator de von Willebrand (vWF), trombospondina (TSP), fibrinogênio (FB) e fibronectina (FN) como intermediários, fazem a célula falciforme interagir com moléculas das células endoteliais como a laminina, glicoproteína Ib (GPIb), integrinas, molécula de adesão celular-vascular (VCAM) e receptor Fc (Fc-R) conforme mostra a figura 6.35. |
||||||||||||||||||||||||||||||||||||||||||
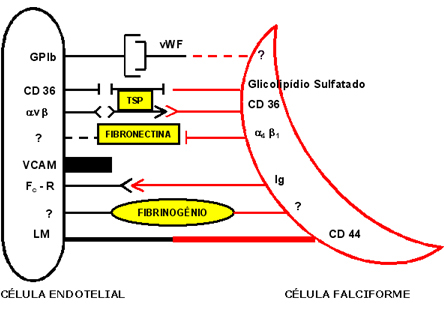 Figura 6.35 - Possíveis interações entre a Célula Endotelial e a Célula Falciforme. GPIb (glicoforina Ib); vWF (Fator de von Willebrand); VCAM (molécula de adesão célula vascular); FcR (receptor Fc); LM (laminina); Ig (Imunoglobulina); TSP (trombospondina). Em amarelo estão representados os fatores plasmáticos que interagem com a célula endotelial e a célula falciforme. |
||||||||||||||||||||||||||||||||||||||||||
A gravidade da doença foi relacionada com a capacidade dos eritrócitos falciformes aderirem à camada superficial das células endoteliais. A adesividade dos eritrócitos falciformes permanece inalterada durante os episódios de dores agudas, exceto quando influenciada por qualquer mudança nas subpopulações de eritrócitos. Ao finalizar este item, é importante destacar que trabalhos recentes demonstraram que há uma formação diferenciada do endotélio de pessoas com doenças falciformes em relação ao endotélio de pessoas com Hb AA, e certamente este fato é determinante para a adesividade das células falciformes. |
||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||