Autores:pppp Paulo Cesar Noum Paulo Francisco Naoum Júnia Iannella Alves |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A
maioria das variantes estruturais é originada por simples
substituições de aminoácidos, resultantes de
mudanças nas seqüências de nucleotídeos.
As alterações estruturais, com conseqüências
nas atividades físico-químicas da molécula,
estão na dependência da extensão do processo
mutacional e dos locais em que esses ocorrem. Dessa forma, as hemoglobinas
variantes podem originar-se por: a) Substituição de um aminoácido por outro, de características diferentes, na superfície externa (figura 5.2) da molécula. Pode ocorrer também a substituição de dois aminoácidos por outros dois, em uma mesma cadeia, sendo, entretanto, condição muito rara. As substituições de aminoácidos na superfície externa, com exceção feita às Hb S, Hb C e Hb E, não produzem alterações significantes no funcionamento da molécula. Nesse grupo estão cerca de 500 tipos de Hb variantes não patológicas (ex.: Hb O, Hb J, Hb I, Hb N, Hb D, etc.). Substituições de aminoácidos na superfície interna da molécula, envolvendo resíduos polares e não-polares, tem ocorrido preferencialmente nos locais invariantes da molécula, incluindo aqueles que fazem parte do "pacote" do grupo heme, cuja principal função é protegê-lo da entrada de água, bem como dos aminoácidos que participam dos contatos a1 b1. Qualquer substituição na superfície interna causa instabilidade molecular, geralmente iniciando-se pela oxidação do grupo heme com a formação excessiva de metaemoglobina e precipitação da globina instável. Citologicamente é possível observar a precipitação intra-eritrocitária da globina instável por meio da presença de corpos de Heinz. Nesse grupo estão cerca de 200 tipos diferentes de Hb Instáveis (figura 5.2). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
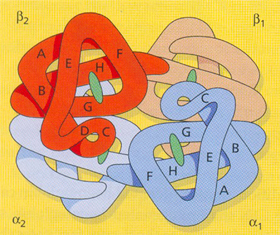 Figura 5.2: Estrutura do tetrâmero da Hb A com identificações de suas subunidades a1, a2, b1e b2. Superfície externa: corresponde aos aminoácidos das regiões A, B e F nas globinas alfa e beta. Superfície interna: D e C. Grupo heme: G e H. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
b) Substituições de aminoácidos que participam dos contatos a1b2, das ligações químicas com o 2,3 DPG, e do resíduo histidina C-terminal da cadeia beta promovem a formação de hemoglobinas variantes com alterações na sua afinidade pelo oxigênio. São cerca de 50 tipos as Hb variantes com afinidade aumentada ou diminuída por oxigênio. As que têm afinidade aumentada se destacam por eritrocitoses, enquanto aquelas com afinidade diminuída manifestam-se notadamente por anemia hemolítica (figura 5.2). c) Substituição dos resíduos de histidina distal ou proximal, que estão ligados ao grupo heme (figura 5.2), causam anormalidades que se caracterizam pela oxidação espontânea e contínua do ferro, com formação excessiva de metaemoglobina, fato que dão origem às hemoglobinas variantes do tipo M (Hb M). Os portadores de Hb M são sempre cianóticos, com ou sem anemia. d) Adição de um ou mais aminoácidos ao último aminoácido (C-terminal) das globinas alfa e beta, tornando-as longas e manifestando-se como fenótipos talassêmicos alfa e beta (ex.: Hb Tak). e) Fusão entre duas cadeias de globinas diferentes, em especial das cadeias delta-beta que resultam na formação da hemoglobina variante conhecida por Hb Lepore. A fusão inversa, ou seja, beta-delta é conhecida por Hb anti-Lepore. Outras fusões têm sido descritas na literatura e todas essas ocorrências se devem ao "crossing-over" desigual no pareamento dos cromossomos 11. Assim, somam-se atualmente perto de 700 variantes estruturais, poucas delas associadas com manifestações clínicas e alterações hematológicas, que podem ser agrupadas em: - hemoglobinas de agregação; - hemoglobinas sem alterações fisiológicas; - hemoglobinas instáveis; - hemoglobinas com alterações funcionais; - hemoglobinas com fenótipos talassêmicos. As hemoglobinas de agregação
formam tactóides e cristais, com repercussões clínicas
e laboratoriais variáveis. As hemoglobinas S e C participam
desse grupo. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 5.1: Exemplos de hemoglobinas variantes estruturais e seus efeitos fisiopatológicos.
(1) Há vários tipos de hemoglobinas instáveis; (2) Há vários tipos de hemoglobinas com afinidade aumentada por O2; (3) Há poucos tipos de hemoglobinas com afinidade diminuída por O2; (4) Há vários tipos de variantes de Hb A2 por mutação na globina delta; (5) Há vários tipos de variantes de Hb Fetal, somente detectáveis em sangue de recém-nascidos. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 5.2: Doenças causadas por hemoglobinas variantes estruturais.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hemoglobinas variantes comuns no Brasil
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
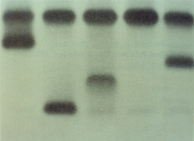
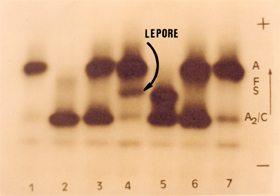 Figura 5.3: Eletroforese alcalina de hemoglobina em acetato de celulose. (1) Hb AA; (2) Hb CC; (3) Hb AC; (4) Hb A + Lepore; (5) Hb SC; (6) Hb AC e (7) Hb A + A2 Aumentada. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Entre as hemoglobinas variantes, o genótipo heterozigoto da Hb C, ou Hb AC, é o segundo mais prevalente após a Hb AS na população brasileira, variando entre 0,3% a 1,0%. A homozigose da Hb C (ou Hb CC) é rara e é caracterizada por anemia hemolítica de intensidade variável, com evidências clínicas de cansaço, fraqueza e, eventualmente, esplenomegalia. Laboratorialmente a hemoglobina total oscila entre 9 e 12g/dl, hematócrito entre 30 e 40%, leve a moderada reticulocitose (3 a 7%) e no esfregaço sangüíneo há muitas células em alvo. Eletroforeticamente, a homozigose da Hb C tem concentração de 98% desta hemoglobina, em pessoas com idade superior a seis meses. Outras condições associadas de Hb C com manifestações clínicas são a doença falciforme por Hb SC, que será apresentada no item 6 (Doença Falciforme), e a interação entre Hb C e talassemia beta, ou Hb C/Tal. beta cuja avaliação eletroforética se torna visível pela Hb CF (figura 5.4). Diferentemente da Hb CC, que não se detecta Hb Fetal com níveis acima do normal, na Hb C/Tal. beta a Hb Fetal geralmente está elevada (>5%). Nesses casos as evidências clínicas são marcadas por palidez, cansaço e esplenomegalia. Laboratorialmente a anemia é moderada (Hb: 9 – 10g/dl) do tipo microcítica, hipocrômica, muitas células em alvo e reticulocitose acima de 5%. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
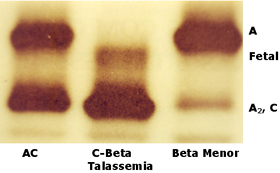
Figura 5.4: Eletroforese em acetato de celulose com pH alcalino (pH 8,6) mostrando da esquerda para a direita: Hb AC, Hb CF (C/Tal. beta) e Hb A + A2 aumentada. Observar que a Hb Fetal tem concentração acima de 5%. A avaliação quantitativa da Hb Fetal pode ser efetuada por densitometria da eletroforese, ou por dosagem bioquímica de Hb Fetal. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hemoglobina D (ou Hb D) – A Hb D é uma hemoglobina variante que apresenta a mesma mobilidade da Hb S em eletroforese de pH alcalino. É separável da Hb S por eletroforese em agar pH ácido (pH 5 a 6), e também por não se insolubilizar em soluções redutoras de oxigênio. Quando associada à Hb A, a heterozigose de Hb AD, o portador é totalmente assintomático, e a fração anormal constitui entre 30 e 50% da hemoglobina total. A prevalência de portadores de Hb AD no Brasil é por volta de 1 caso para cada 5 mil pessoas analisadas. Casos de homozigoses de Hb D (Hb DD) são raríssimos, e podem estar associados a discreto grau de anemia (Hb: 10,5 – 12,0 g/dl). Para estabelecer o diagnóstico de homozigose deve-se excluir cuidadosamente, por estudos familiares, a interação da Hb D com a talassemia beta. Na Hb D/Tal. beta é comum evidenciar no hemograma anemia microcítica e hipocrômica, com hemoglobina total variável de 9,5 a 12 g/dl, VCM abaixo de 77 fl e HCM também abaixo de 27 pg. Situação que oferece dificuldade no diagnóstico laboratorial ocorre quando há associação entre hemoglobinas S e D, ou Hb SD caracterizando um dos tipos que compõe o grupo das doenças falciformes, apresentado no item 6. Neste caso específico, a eletroforese de hemoglobina em pH alcalino não diferencia o genótipo SS da SD, bem como o teste de falcização que é positivo em ambos. O teste mais adequado para a diferenciação é a eletroforese em agarose de pH ácido: a Hb S é mais lenta que a Hb D, que se posiciona igual à Hb A (figura 5.5). Após ter sido descrita em 1953, várias outras hemoglobinas que se posicionavam como a Hb D (e não falcizavam) foram estruturalmente diferenciadas conforme o tipo de substituição de aminoácidos. Assim surgiram as Hb D Los Angeles ou Punjab (a mais freqüente entre todas as hemoglobinas variantes que migram na mesma posição da Hb S), Hb D Iran, Hb D Ibadan, etc. todas na posição de Hb S. Atualmente, há dezenas de hemoglobinas variantes que migram na posição de S ou D e que foram denominados por local de origem e que são diferenciadas em eletroforeses em agar ácido, isofocalização, HPLC, e por biologia molecular. A tabela 5.3 mostra a relação de hemoglobinas que migram na mesma posição de Hb S em pH alcalino, e suas diferenciações estruturais. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
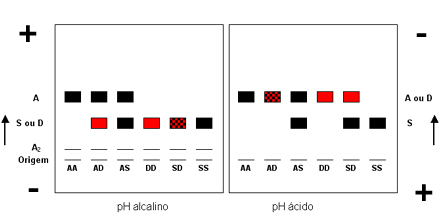 Figura 5.5: Mapa representativo dos principais genótipos de Hb D comparados com Hb A e Hb S, em eletroforeses de pH alcalino e ácido. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabela 5.3: Relação das principais hemoglobinas variantes que migram na posição de Hb S em eletroforese de pH alcalino, que são raríssimas e menos freqüente que Hb D Los Angeles.
* Duplas mutações na globina beta, além das mutações apresentadas todas tem a mutação da Hb S (b6 Glu ® Val). ** Posição eletroforética similar. ¹ Entre Hb A e Hb S. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hemoglobina G (ou Hb G) – É um grupo de hemoglobinas variantes que migram pouco atrás da Hb S nas eletroforeses alcalinas em acetato de celulose e agarose. A diferenciação se faz por meio de eletroforese em agarose ácida. Entre as hemoglobinas do tipo G destacam Hb G Audhali (a 23 Glu ® Val), Hb G Waimanalo (a 64 Asp ® Asn), Hb Filadelfia (a 68 Asn ® Lis), Hb G Galveston (b 43 Glu ® Ala), Hb G Copenhagen (b 47 ® Asn), Hb G Accra (b 79 Asp® Asn), entre outras. A Hb G Filadélfia é a mais freqüente entre todos os tipos de Hb G, e em especial no Brasil e USA pois sua origem é africana. Tem baixa prevalência na população brasileira (cerca de 1: 15000), porém, por ser comum entre pessoas de descendência africana e se situar eletroforeticamente na região próxima da Hb S em eletroforese alcalina, sua avaliação é sempre importante. Por ser a Hb G Filadélfia uma mutante de globina alfa, geralmente afetando um dos quatro genes alfa, sua concentração é quase sempre abaixo de 25%; além disso, quando em heterozigose com a Hb A (Hb AG), é possível separar as seguintes hemoglobinas, conforme mostra a figura 5.6: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
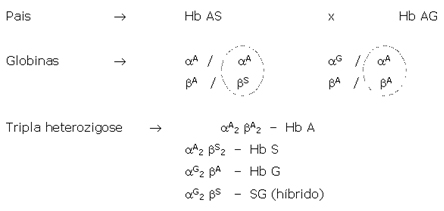
Os portadores de Hb AG e Hb GG são assintomáticos, entretanto é possível a ocorrência da tripla heterozigose entre hemoglobinas A, S e G Filadélfia, proveniente de pais com Hb AS e Hb AG, conforme mostra o esquema a seguir: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A tripla heterozigose forma um quarto
produto híbrido a Hb SG, conforme mostra a figura 5.7.
Hemoglobina Lepore (ou Hb Lepore) – A Hb Lepore é uma hemoglobina variante, com migração similar à Hb S (ver tabela 5.3), causada por um pareamento desigual do cromossomo 11 durante a meiose. Como conseqüência dessa desigualdade na posição das cromátides irmãs do cromossomo 11, o "cross-over" entre elas promove a fusão de uma parte do gene delta com outra do gene beta, formando um gene híbrido delta-beta, além dos genes delta, gama e beta. Esse gene híbrido delta-beta seqüência globinas em que a parte inicial é formada por aminoácidos da globina delta e a parte final por aminoácidos da globina beta, conforme mostra o esquema a seguir: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
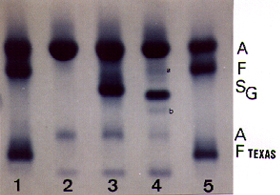
Figura 5.6: Eletroforese de hemoglobina em agarose com pH alcalino, com destaque para as mobilidades de Hb S e Hb G Filadélfia. (1) e (5) sangue de recém-nascido com Hb A + Hb Fetal + Hb Fetal Texas (mutante); (2) Hb AA; (3) Hb AS; (4) Hb AG, com Hb G Fetal (a) e Hb G2 (b). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
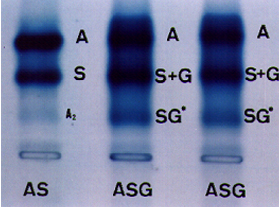 Figura 5.7: Eletroforese de hemoglobina em agarose com pH alcalino, com destaque para a tríplice heterozigose (Hb ASG). A Hb SG* é uma forma híbrida proveniente da combinação entre aG2 e bS2. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Dependendo do local em que ocorre a fusão delta-beta a Hb Lepore pode originar pelo menos três sub-tipos: Lepore Boston, Lepore Baltimore e Lepore Holanda. Todas apresentam as mesmas características laboratoriais e eletroforéticas, sendo diferenciadas por estudos de composição peptídica da fusão delta-beta. A concentração da Hb Lepore em eletroforese alcalina é variável entre 5 e 15%, com Hb A2 normal ou diminuída; algumas vezes a Hb Fetal pode estar elevada. Nesses casos, o quadro hematológico laboratorial é típico de talassemia beta menor com aniso-poiquilocitose, microcitose e hipocromia. Situação mais grave ocorre na homozigose da Hb Lepore, com quadro clínico e laboratorial semelhante à talassemia maior ou intermédia. A eletroforese de hemoglobina alcalina nas pessoas com Hb Lepore homozigota mostra a presença de Hb Fetal e Hb Lepore com concentrações elevadas. A figura 5.8 ilustra as situações da Hb Lepore heterozigota e homozigota. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
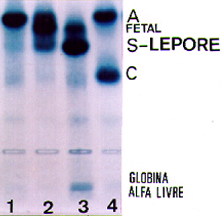
| Figura 5.8: Eletroforese de hemoglobinas em agarose alcalina mostrando em (1) Hb Lepore heterozigota com concentração de 5%; (2) Hb Lepore homozigota, com Hb Fetal ± 70%, Hb Lepore ± 15%, Hb A (transfundida) ± 15%, e traços de Hb A2; (3) Hb SF de um paciente com Hb S/b0 talassemia, com globinas alfa livre; (4) Hb AC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||