Análises Eletroforéticas
de Hb Variantes Raras
Autor: Paulo Cesar Naoum |
||||||||||||||||
1. HEMOGLOBINAS
A e A2
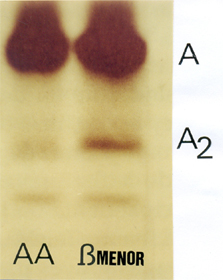
As hemoglobinas são proteínas complexas de estrutura quaternária que desempenham o transporte de oxigênio e participam do equilíbrio ácido-básico. Cada eritrócito contém, em média, cerca de 300 milhões de moléculas de hemoglobinas (ex.: HCM = 30pg). Após o sexto mês de vida de uma pessoa com síntese normal para hemoglobinas é possível identificar com clareza as Hb A e Hb A2. Essa identificação se faz por métodos de fracionamento protéico, dos quais o mais conhecido é a eletroforese de acetato de celulose em pH alcalino. Além das Hb A e Hb A2 pode aparecer a Hb Fetal, cuja concentração atinge no máximo 1%; na maioria das vezes está ausente. A concentração da Hb A é variável de 96 a 98% e a Hb A2 de 2 a 4%. Uma pessoa com concentrações normais para hemoglobinas A, A2 e Fetal é identificada por Hb AA. Quando a Hb A2 está aumentada (>4% até 8%) é sugestivo de que o portador tenha talassemia beta menor. Para esses casos há necessidade de comprovações clínicas e hematológicas. 2. Hb VARIANTES COMUNS E RARAS
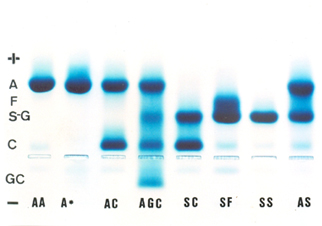
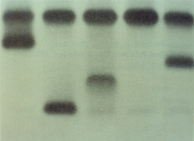
Esse grupo de oito análises mostra hemoglobinas normais (Hb AA), Hb variantes comuns (AC, SC, SF, SS e AS), Hb variante rara (AGC) e um caso em que a deficiência de ferro diminui a Hb A2 (A*). Análise específica de cada amostra: Hb AA – Padrão normal com Hb A (96,7%) Hb A2 (3,3%). Hb A* – Hb A2 diminuída (0,6%) em paciente com ferropenia. Hb AC – Heterozigose de Hb C, prevalente em 0,5% na população brasileira. A Hb C é uma mutante de globina beta (b 6 Glu ® Lis) e tem origem africana. O portador é assintomático. Hb ACG – Situação muito rara de dupla heterozigose. A Hb C, como se viu acima, é uma mutante de globina beta, enquanto a Hb G é mutante de alfa (a 68 Asn ® Lis). É provável que os pais do paciente sejam portadores de Hb AC e Hb AG, e a herança ocorreu da seguinte forma:
Hb SC – Dupla heterozigose que caracteriza a doença falciforme SC. O paciente padece de anemia hemolítica moderada a grave. Prevalência no Brasil de 1:20.000. Hb SF – Nesse caso com a Hb Fetal elevada (22,5%) associada à Hb S (73,4%) e Hb A2 (4,1%), com VCM e HCM diminuídos, trata-se da interação de Hb S com talassemia beta, ou Hb S/tal. beta. Prevalência na população brasileira 1:30.000. O paciente padece de anemia moderada a grave. Hb SS – Anemia falciforme, ou homozigose de Hb S. A anemia é quase sempre acentuada. A prevalência na população brasileira é de 1:10.000. Hb AS – Traço falciforme. O portador é assintomático e a prevalência na população brasileira é de 2,1%. Na Bahia a prevalência varia entre 5 a 10% devido ao predomínio da população negra, enquanto que em Santa Catarina a prevalência é de 0,5% devido à marcante colonização européia. 3. FAMÍLIA COM Hb S/TALASSEMIA BETA
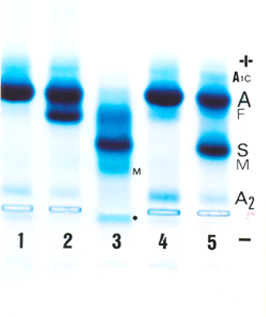
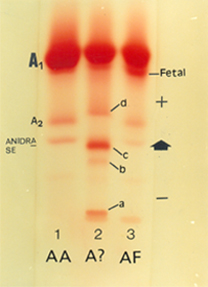
A interação entre Hb S e talassemia beta, ou Hb S/tal. beta, como mostra a análise de nº 3 na eletroforese, é caracterizada clinicamente como doença falciforme. O estudo eletroforético revela, quase sempre, o perfil de Hb SF, com concentração de Hb F (ou Fetal) entre 5 e 20%. Geralmente os pais são assintomáticos, como nesse caso; a análise nº 4 é o pai, com talassemia beta menor (Hb A2: 5,3% e Hb Fetal: 3,1%, ambas elevadas) e a mãe (nº 5) é portadora de traço falcêmico. A análise nº 1 é um padrão normal para hemoglobina (Hb AA) enquanto que a de nº 2 é um padrão com Hb Fetal elevada, obtida de um recém-nascido (Hb AF). No presente caso de Hb S/tal. beta (nº 3) é possível observar elevação de meta Hb S (identificada por M) e por globinas alfa livres (comuns na talassemia beta) e identificada por (*). O desempenho qualitativo dessa eletroforese foi excelente e permitiu a identificação do grupo de hemoglobinas glicadas (A1a, A1b, A1c e A1d) identificadas como A1c. 4. Hb H E TALASSEMIA ALFA
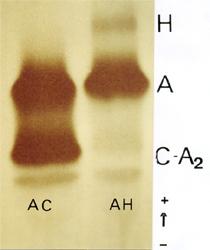
A talassemia alfa é a mais prevalente alteração genética da hemoglobina em todas as populações mundiais. Sob o ponto de vista clínica há quatro categorias de talassemia alfa: mínima, menor, intermédia e maior. A caracterização laboratorial da talassemia alfa se faz pela pesquisa eletroforética da Hb H. O presente caso é de talassemia alfa mínima em que a concentração de Hb H é 2,5%, o eritrograma do paciente apresentava valores normais com discreta alteração na morfologia eritrocitária. No Brasil, a tal. alfa mínima tem prevalência de 20 a 30%. Há países em que a prevalência de talassemia alfa mínima chega a 70%. Na presente eletroforese confrontamos a Hb AH (tal. alfa mínima) com a Hb AC. Observe que a Hb A2 do paciente com Hb AH está diminuída – é um fato constante na talassemia alfa a diminuição de Hb A2. Maiores informações sobre talassemia alfa você encontrará no link artigos científicos do nosso site, ou no www.cdalab.com.br 5. Hb INSTÁVEL (Hb KOLN)
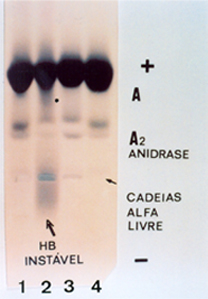
(1) Talassemia beta menor com a Hb A2 aumentada (A2 > 4%). Eritrograma com microcitose e hipocromia (Hb: 13,3g/dL; VCM: 68 fL; HCM: 26 pg) (2) Hb Instável em paciente do sexo feminino, 32 anos, branca. Observar um rastro identificado por (*), além da visível fração espalhada atrás do ponto de aplicação e que é composta por cadeias livres de globinas alfa. Trata-se da Hb Instável denominada por Hb Koln (mutação no resíduo 98 da globina beta: Valina ® Metionina). O eritrograma evidenciou anemia moderada (Hb 9,8 g/dL), VCM: 96 fL com macrocitose e policromasia devido à reticulocitose de 33%; hipocromia (HCM: 24 pg); corpos de Heinz em 70% dos eritrócitos e metaemoglobina aumentada (5,5%). Observar que a Hb A2 do paciente com Hb Koln apresenta-se em posição diferente aos das amostras (1) e (4). É importante destacar que o hemolisado para eletroforese da hemoglobina Instável foi feito com a saponina a 1%, porisso foi possível visualizar as cadeias alfa livres. (3) Sangue do paciente com Hb Instável. O hemolisado foi feito com clorofórmio que destrói as evidências da Hb Koln (*) e das cadeias alfa livre. Apenas a Hb A2 conserva a posição deslocada em relação à Hb A2 das amostras (1) e (4). (4) Amostra de hemoglobina normal – Hb AA 6. HEMOGLOBINA INSTÁVEL POLIMERIZADA
(1) Hb AA – hemoglobinas normais (Hb A ou Hb A1 = 97%; Hb A2 = 3%). Obs.: Hb A1 é a forma antiga de designar a Hb A. (2) Hb A + Hb Instável – Paciente com 12 anos, masculino, branco, apresentou anemia moderada (Hb: 8,6 g/dL), morfologia eritrocitária com anisocitose e poiquilocitose, com destaque para “células mordidas”, reticulocitose de 17%, presença de corpos de Heinz em 50% dos eritrócitos, metaemoglobina elevada (6,7%) e bilirrubina total e indireta aumentadas. As frações de hemoglobinas (a), (b) e (c) são polímeros da Hb Instável. É muito comum a Hb A2 (d) das hemoglobinas instáveis apresentar-se discretamente acima da Hb A2 normal. (3) Hb AF – Hb Fetal aumentada, com concentração de 5%, em paciente portador de talassemia beta menor. (GV: 5 x 106/mm3; Hb: 10,3 g/dL; Ht: 37%; VCM: 74 fL; HCM: 20,6 pg; RDW: 18) 7. Hb KORLE-BU ASSOCIADA ÀS TALASSEMIA ALFA, BETA+ E DELTA
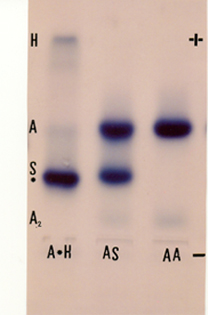
A Hb Korle-Bu é uma variante rara, mutante de globina beta (b 73 Asp ® Asn), e de origem africana. Na eletroforese se posiciona discretamente mais lenta que a Hb S. O presente caso pertencia a um paciente com anemia microcítica e hipocrômica (Hb: 10,1 g/dL, VCM: 72 fL e HCM: 20,2 pg). Todos os testes para Hb S foram negativos. Ao submetermos a amostra hemolisada com saponina a 1% observamos que a Hb anormal (Korle-Bu) era pouco mais lenta que a Hb S, apresentava Hb H (8,5%), a Hb A2 estava ausente e havia cerca de 5,0% de Hb A. Na eletroforese se trata da primeira análise à esquerda e identificada por Hb A*H; o * foi colocado antes da análise molecular que revelou ser Hb Korle-Bu. Um dos pais era heterozigoto para Hb Korle-Bu e Hb H (Hb A/ Korle-Bu/ tal. alfa) e outro tinha b+/d0 talassemia menor. As outras duas análises (Hb AS e Hb AA) foram colocadas na eletroforese como padrões de avaliação. 8. ELETROFORESE DE FOCALIZAÇÃO ISOELÉTRICA PARA HEMOGLOBINAS
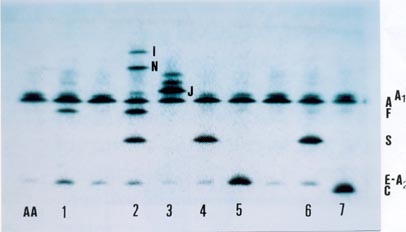
Esta eletroforese pode ser feita em gel de agarose ou poliacrilamida. Mistura-se a um desses géis um composto químico conhecido por anfolina e que é capaz de dispor a composição desses géis em faixas específicas de pH variáveis entre pH 1 a pH 14. Ao aplicar amostras de hemoglobinas nesses géis, à medida que a hemoglobina A, S, Fetal, C, I, etc, se desloca, encontra seu ponto isoelétrico idêntico ao do pH da faixa, e assim ocorre a precipitação da hemoglobina (focalização). Observe nesta foto a nítida separação das hemoglobinas A, F, S, C, E, A2 I, N, J e A1c. A amostra identificada pelo número 1 é de um caso de talassemia beta menor com elevações de Hb F e Hb A2; a amostra nº 2 é um padrão com Hb A, F, S, I e N; a de nº 3 é de Hb AJ e duas outras variantes não identificadas; a de nº 4 e nº 6 são de Hb AS (traço falciforme); a de nº 5 é Hb AE e a de nº 7 é Hb AC. 9. Hb PORTO ALEGRE (Beta 9 Serina » Cisteína)
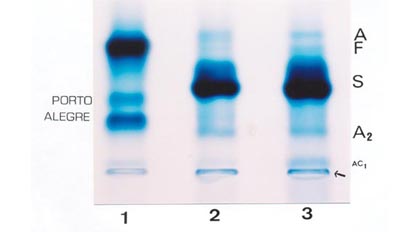
A mutação que deu origem à Hb Porto Alegre é capaz de formar dois ou mais polímeros desta variante à medida que o hemolisado se torna envelhecido. Observe na aplicação 1 que a Hb Porto Alegre tem duas frações (polímeros), uma na posição próxima da Hb C (mais concentrada) e outra na posição similar de Hb S. É um caso de heterozigose, pois está associada à Hb A. As aplicações 2 e 3 são amostras de um paciente com anemia falciforme (Hb SS) com discreta concentração de Hb A – resquício de uma transfusão de sangue – e traços de Hb Fetal. 10. Doença de Hb H e outras variantes comuns
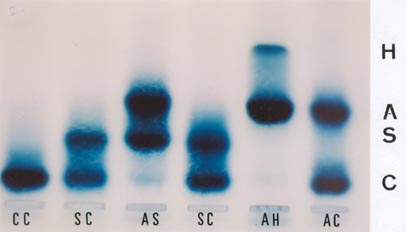
A doença de Hb H se caracteriza quando três ou quatro genes de globina alfa estão com suas sínteses diminuídas em relação à globina beta. Algumas vezes três genes alfa podem ter bloqueio total de síntese ou os quatro genes alfa podem ter sínteses parciais de globina alfa. Além de anemia com graus moderado a grave observável no eritrograma, as eletroforeses de hemoglobinas em pH alcalino ou pH neutro permitem a identificação da Hb H. O presente caso em eletroforese alcalina em gel de agarose mostra Hb H com concentração de 12%; trata-se de um paciente com a doença de Hb H (Hb: 9,6 g/dL; VCM: 67 fL; HCM: 19 pg). As outras variantes são Hb CC (homozigose), Hb SC (doença falciforme), Hb AS (traço falciforme) e Hb AC (heterozigose). 11. Hb Instável: Santa Anna (Beta 88 Leu – Pro) – Eletroforese
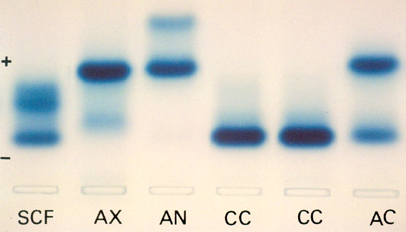
A eletroforese em agarose alcalina mostra da esquerda para a direita as seguintes hemoglobinopatias: a) Hb SCF – sangue de criança com quatro meses de idade portadora de doença falciforme por Hb SC e com Hb Fetal elevada devido à idade. b) Hb AX – A representação X é para a hemoglobina instável denominada por Santa Anna. A fração anormal é difusa e situa-se entre a Hb S e Hb C. O paciente apresentou na oportunidade anemia moderada (Hb 8,8 g/dL, VCM: 78 fL e HCM: 26 pg), presença de corpos de Heinz (próxima foto, nº 12) e teste de instabilidade térmica positiva (foto nº 13). c) Hb AN – A Hb N é uma variante de globina beta (95 Lis-Glu) que migra mais rápido que a Hb A e não é patológica. d) Hb CC – Homozigose da Hb CC, com discreta anemia microcítica e hipocrômica (Hb: 10,7 g/dL; VCM: 68 fL e HCM: 24 pg). e) Hb CC – idem f) Hb AC – Heterozigoto da Hb C (AC) sem conseqüência clínica. 12. Hb Instável: Santa Anna (Beta 88 Leu – Pro) – Corpos de Heinz
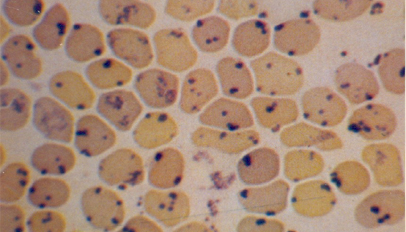
A mutação do aminoácido leucina por prolina ocorre na região interna da molécula de hemoglobina, justamente onde há os contatos químicos entre as globinas alfa e beta. Com a mutação esses contatos ficam fracos, a molécula se desestabiliza e ocorre a precipitação da globina beta mutante. Os precipitados da globina beta mutante dentro dos eritrócitos formam agregados protéicos que se coram e são conhecidos por corpos de Heinz. 13. Hb Instável: Santa Anna (Beta 88 Leu – Pro) – Precipitação
O teste de precipitação da Hb Instável é feito à temperatura de 50ºC por 60 minutos. A Hb Santa Anna precipitou aos 10 minutos – comparar com o padrão – e a globina beta se sedimentou no fundo do tubo aos 30 minutos. Este é o teste de instabilidade térmica para hemoglobinas instáveis. |
||||||||||||||||
 |
||||||||||||||||
|
||||||||||||||||